Epilepsy with myoclonic-atonic seizures (EMAS), also known as Doose syndrome, is characterized by the presence of myoclonic-atonic seizures (MAS) in an otherwise normal child who may have a history of febrile and/or afebrile seizures [1]. Doose syndrome was previously classified as cryptogenic or idiopathic epilepsy with primary generalized minor seizures with Lennox-Gastaut syndrome (LGS) in the 1970s and 1980s [1,2]. However, research eventually distinguished Doose syndrome and LGS as different diseases. The recommended therapies for Doose syndrome include anti-seizure medication (ASM) and diet therapy, such as ketogenic diet (KD) and modified atkins diet (MAD). ASMs such as valproic acid, levetiracetam, clobazam, lamotrigine, ethosuximide, and topiramate are known to be effective. However, it has been reported that pharmacoresistance occurs in about 90% of cases. When diet therapy is applied to patients with Doose syndrome, it is known that seizure reduction of more than 50% is achieved in about 80% of patients [3].
Although there is no consensus on the most suitable treatment because Doose syndrome is mostly drug-resistant, diet therapy is by far the most effective. However, the application of KD and MAD in pediatric patients is somewhat difficult due to dietary discomfort and gastrointestinal troubles [3-5]. Recently, growing evidence has shown that low glycemic index treatment (LGIT) is as good as the classic KD or MAD in terms of efficacy for drug-resistant epilepsy [4,6]. Herein, we report the case of a patient with Doose syndrome who showed improvement in clinical seizures and electroencephalogram (EEG) after LGIT. This study was approved by the Institutional Review Board of the Gangnam Severance Hospital, Yonsei University College of Medicine. Informed consent for this retrospective study was waived by the board (3-2022-0135).
An 8-year-old boy with no related medical history and with normal development had his first seizure when he was 5. At that time, he complained of sudden fall-like events and frequent myoclonus. The main seizures were MAS, with absence seizures occurring frequently. In addition, clonic seizures of both extremities with impaired awareness often lasted for 3 to 5 minutes. There was no past history of febrile seizures and no family history of seizures. Five months after his first seizure, the same seizures happened again, so he was treated with valproate acid from the regional tertiary hospital.
As the patient complained of memory loss and seizure recurred once every 2 weeks, he visited our hospital outpatient clinic. His magnetic resonance imaging (MRI) was normal and his awake EEG demonstrated frequent multifocal sharp wave discharges. His memory loss was considered as an additional symptom of worsening of convulsions and EEG findings. The addition of oxcarbazepine and dosage increment in valproate acid led to improvement in memory loss and seizure free status for 5 months. Doose syndrome was not suspected at this time.
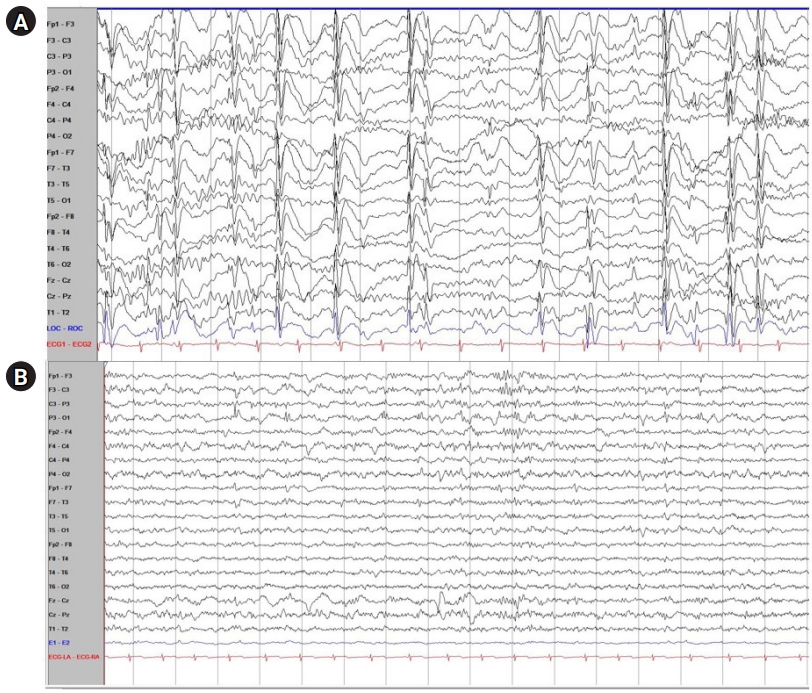
However, myoclonic and atonic seizure recurred at the age of 7, and his follow-up EEG showed frequent multifocal sharp wave discharges and very frequent generalized sharp and slow wave (GSSW) discharges. In his video-EEG, the same result as his follow-up EEG was obtained (Fig. 1A). Considering his normal development before seizure onset, age when the seizures initiated, type of seizure, and his EEG and MRI findings, the patient was diagnosed with Doose syndrome. His medication changed after diagnosis: the patient was additionally prescribed clobazam while oxcarbazepine was stopped.
Two months after he was diagnosed with Doose syndrome, there was no significant improvement in the intensity and frequency of his seizures and therefore, the patient started LGIT. He showed high compliance with LGIT and there was no side effect during his admission. One month after the LGIT initiation, GSSWs were no longer observed in the follow-up EEG (Fig. 1B). Thus, the patient was in a better condition without any clinical seizures. Furthermore, LGIT was effective without causing any gastrointestinal trouble after discharge. In his developmental assessment report 3 months after starting LGIT, his social age was 8.5 years old and his social index was 108.55, which is rather average at his age. Although the objective developmental status of the patient before the start of LGIT was not investigated, the patient and caregiver reported that the patient's concentration and memory gradually improved after LGIT. We plan to continue the treatment by maintaining LGIT and tapering ASM with routine EEG, laboratory findings, and nutritional checkups.
At present, the most updated International League Against Epilepsy (ILAE) diagnostic manual of Doose syndrome includes: (1) the onset of epilepsy between 6 months and 6 years of age (peak: 2 to 4 years); (2) mostly normal development and cognition before the onset of epilepsy without clear neurological deficit; (3) mandatory MAS; (4) presence of generalized spike-waves discharged at 2 to 3 Hz without persistent focal spike discharges; and (5) exclusion of other myoclonic epilepsy syndromes [1]. For the diagnosis of Doose syndrome, it is necessary to differentiate diseases such as LGS, Dravet syndrome, and developmental epileptic encephalopathy with spike-and-wave activation in sleep [7]. In recent years, genetic and structural etiologies have been identified as potentially causative [8].
As Doose syndrome is mostly pharmaco-resistant, diet therapy is thus so far the most potent treatment. The KD is a high-fat, low-carbohydrate, and adequate-protein regimen utilizing strictly controlled amounts and ratios of fat to a combination of protein and carbohydrates [6,9]. It mainly includes classic KD, medium-chain triglyceride diet (MCTD), MAD, and LGIT [6,9,10]. MCTD is used to replace long-chain fatty acids of the classical KD with up to 60% of calories [9]. MAD is less restrictive with low carbohydrates restricted to 10 to 20 g/day. LGIT consists of a more liberal carbohydrate intake of up to 40 to 60 g/day with a glycemic index of <50 to prevent hypoglycemia [6,9].
Not all mechanisms of reduction in seizure induced by KD are fully elucidated, but there are two reasonable hypotheses. First, the ketone bodies produced by KD increase the inhibitory neurotransmitters, activate the potassium channel, and enhance brain energy production, thus increasing the seizure threshold [6]. Moreover, fatty acids and polyunsaturated fatty acids (PUFA), especially n-3 PUFA, activate peroxisome proliferator-activated receptors, which improve neuronal function, regulate ion channels that leads to neuronal hyperpolarization, and upregulate uncoupling proteins thereby decreasing reactive oxygen species production [6]. Such mechanisms eventually bring about an anticonvulsant effect. Despite the high efficacy of KD, there could be several adverse effects such as gastrointestinal disturbance, dyslipidemia, growth failure, kidney stone, and rarely respiratory failure and pancreatitis. Therefore, it is often difficult to maintain KD or MAD therapy for pediatric patients [3,6].
Compared with classic KD and MAD, LGIT is less restrictive, much safer, and has fewer adverse events. It is less burdensome for childrenŌĆÖs consumption because LGIT causes less gastrointestinal trouble and it is much easier to educate caregivers since the LGIT composition itself is not as strict as other diets. Although Wiemer-Kruel et al. [5] have emphasized the importance of the MAD as an effective treatment for Doose syndrome, as far as we know, the current study is the first to report the efficacy of LGIT in Doose syndrome. The efficacy of LGIT has been proven to not be inferior to classic KD and MAD, and can be safely and effectively used in epileptic encephalopathies such as Doose syndrome [4].
In summary, we report that among all diet therapies, LGIT can be effective in improving clinical seizure and EEG in patients with Doose syndrome. Since diet therapy such as KD is known to be more effective than multiple ASMs in the treatment of Doose syndrome, a diet therapy that is easier to eat and has fewer side effects may be necessary for pediatric Doose syndrome patients. LGIT seems to be a good alternative in such situation. The findings from this case are expected to be helpful for pediatric neurologists treating patients with Doose syndrome.