Introduction
Acute disseminated encephalomyelitis (ADEM) is an acute or subacute immune-mediated demyelinating inflammatory disorder of the central nervous system (CNS). It is characterized by encephalopathy with multifocal neurological deficits and characteristic magnetic resonance imaging (MRI) findings of widespread demyelination, predominantly involving the white matter of the brain and spinal cord, and mostly occurs after recent viral or bacterial infections [1-4]. It is predominantly diagnosed in children, is usually monophasic, and often resolves within 3 months of treatment. The International Pediatric Multiple Sclerosis Society Group (IPMSSG) made enormous efforts to create consensus clinical and radiological diagnostic criteria for ADEM in 2013 (Table 1) [5]. In addition, the discovery of myelin oligodendrocyte glycoprotein antibody (MOG-Ab) and understanding of its association with recurrent forms of ADEM, such as multiphasic ADEM (MDEM), have made its diagnosis and management possible [6-8]. Despite these advances, the diagnosis is still made based on clinical and MRI findings, and minimal modifications have been made in the treatment protocol over these years. Although the overall prognosis is usually favorable, some patients with ADEM might develop neurological sequelae such as cognitive impairment, motor deficits, visual problems, and epilepsy [9-11]. Relapse can occur in a minority of children with ADEM, although there are gray zones between relapsing ADEM and other neuroinflammatory demyelinating conditions, such as neuromyelitis optica spectrum disorder (NMOSD) and MOG-Ab-associated disorders (MOGAD). This review focuses on the current knowledge and recent advances in the management of pediatric ADEM.
Epidemiology
ADEM can occur at any age, but predominantly develops in childhood and adolescence. Its annual incidence is estimated to be 0.3 to 0.6 cases per 100,000 children with a mean age of 5 to 8 years at presentation [2,4,12]. Previous studies have reported a slight male preponderance, with male-to-female ratios ranging from 1:0.8 to 2.3:1 [13]. Its incidence is highest in winter and spring [12]. It is also more common with increasing distance from the equator, similar to multiple sclerosis (MS) [2]. ADEM is now considered a form of autoimmune encephalitis. It often occurs after infection or, rarely, after vaccination. In 50% to 86% of cases, ADEM is reportedly preceded by an acute infectious illness such as upper respiratory infections, gastroenteritis, and rarely an exanthematous disease due to various agents [1,4,12,14]. Clinical symptoms typically begin within 2 days to 3 weeks after an infectious event [12]. The most frequent associated infections are viral infections, but bacteria or other agents have also been implicated (Table 1) [2,12,14-19]. Pediatric ADEM cases after infection with severe acute respiratory syndrome coronavirus 2 have also been reported [20,21]. Vaccinations have been implicated as a triggering factor for ADEM in 4% to 18% of cases [1]. Almost all vaccines have been implicated; however, no clear causal association has been proven (Table 1). Reportedly, the first vaccination is more likely to trigger ADEM than subsequent vaccinations [22].
Pathogenesis
The pathogenesis of ADEM remains unclear, but may involve an immune-mediated inflammatory process triggered by infection or vaccination in genetically predisposed individuals. Previous studies have proposed that some children with certain human leukocyte antigen subtypes are more likely to develop ADEM [23-25]. Molecular mimicry between antigenic determinants of neurotropic viruses or other causative agents and myelin agents such as myelin basic protein (MBP) and MOG is considered a putative mechanism underlying immune-mediated neuronal injury [2,26,27]. Another theory is non-specific self-sensitization of reactive T cells (bystander activation) against myelin proteins resulting from infectious antigens [28]. This reactivation makes it possible for T cells to migrate through glial limitans and enter the brain parenchyma. The production of cytokines and chemokines by antigen-presenting cells and activated T cells contributes to further recruitment, which boosts migration into the CNS of additional immune cells, such as T cells and macrophages. Breakdown of the blood-brain barrier is also caused by the release of proteases from activated T cells, mast cells, and monocytes. Furthermore, T cells possibly play a secondary role in other inflammatory processes that can cause demyelination and axonal injury [29]. The autoimmune hypothesis is also supported by the fact that anti-MOG-Abs have been identified in both serum and cerebrospinal fluid (CSF) during the acute phase in 36% to 60% of children with ADEM, which progressively declines during the recovery phase [30,31]. High and persistent MOG-immunoglobulin G titers are usually associated with relapsing forms of MOGAD [31-33]. However, it remains uncertain whether anti-MOG-Abs are causative agents or byproducts of extensive myelin degradation in ADEM or other demyelinating diseases.
Clinical/diagnostic perspectives
1. Clinical features
The initial symptoms typically begin within 2 days to 3 weeks of infection or vaccination. The clinical presentation is diverse; however, patients with ADEM typically show non-specific prodromal symptoms, such as fever, malaise, headache, nausea, and vomiting shortly before neurological symptoms or signs. The clinical course is characterized by rapid progression and development of maximum deficits within a few days [12]. Neurological manifestations typically begin with encephalopathy, characterized by altered consciousness, such as irritability, lethargy, coma, and abnormal behavior, which are associated with focal or multifocal neurological deficits depending on the lesions. In addition, they can present with masquerading symptoms or signs, such as fever, seizures, and meningeal irritation, resembling meningoencephalitis. ADEM can affect any part of the brain, spinal cord, or peripheral nerves, meaning that it can present with any type of neurological symptoms. Common neurological symptoms include pyramidal signs, cranial neuropathy, speech impairment or aphasia, sensory deficits, visual disturbances, ataxia, and spinal cord signs (Table 2). Children with anti-MOG-Abs tend to have multifocal neurological symptoms but show complete resolution after steroid treatment [4]. Pediatric intensive care unit (PICU) admission is required in 15% to 25% of children with ADEM, mainly due to respiratory failure, which results from coma, brainstem involvement, and status epilepticus [1,6,34]. Combined central and peripheral demyelination is also common in children. However, a further diagnostic work-up can be performed to screen for other conditions, such as metachromatic leukodystrophy and Krabbe disease in certain cases. ADEM is typically a monophasic condition but can show a relapsing form described as “recurrent” if the lesions are always the same, or “multiphasic” if the space and time of the lesions are widely dispersed. This relapsing form, such as MDEM, appears to be related to high and persistent MOG-Ab titers [4,31,32].
2. Neuroimaging features
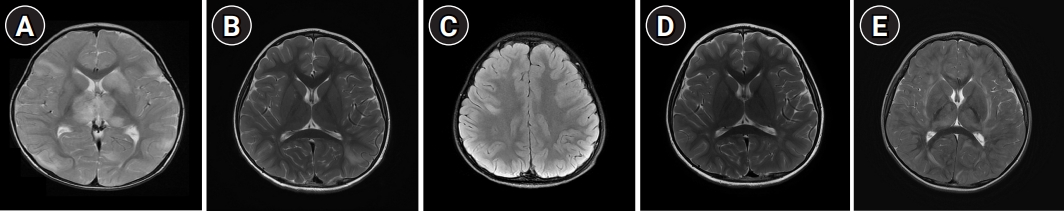
Because there are no specific biomarkers for diagnosis, MRI is still the most sensitive technique for diagnosing ADEM. Brain MRI in the acute phase typically shows diffuse, bilateral, randomly distributed, large (1 to 2 cm), patchy or tumor-like, often heterogeneous, poorly demarcated hyperintense lesions on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences [12,26]. Both white and gray matter are affected. When the deep gray matter is affected, it is more likely to be symmetrical and often involves the thalami and basal ganglia. Apart from these lesions, other patterns have been reported in pediatric ADEM (Table 3) [1,12,35]. Fig. 1 demonstrates various patterns of MRI lesions in pediatric patients with ADEM. Brain MRI may show subtle or no abnormalities in the early phase of ADEM. This type of delay should be taken into consideration if the case is clinically suggestive of ADEM, but the initial MRIs do not show any abnormal findings [36,37]. Spinal cord involvement has been reported in 18% to 80% of children with ADEM, and longitudinally extensive transverse myelitis (LETM) involving at least three vertebral body segments in length has been detected in 60% to 100% of patients [1]. Children with anti-MOG-Ab positive ADEM are at a higher risk of LETM [32]. Gadolinium enhancement is reported in 18% to 50% of patients [1,6]. There have been conflicting results regarding MRI diffusion patterns. However, no restricted diffusion on diffusion-weighted imaging and apparent diffusion coefficient map with high values consistent with vasogenic edema were found in 75% of patients [1]. Verifying the resolution of lesions in serial MRI studies can play a crucial role in bolstering the initial diagnosis of ADEM.
3. Laboratory findings
There are no laboratory tests specific to ADEM. Many tests are useful in ruling out other neurological conditions. CSF findings are usually non-specific, including mild pleocytosis with lymphocyte predominance and slightly elevated protein levels. Oligoclonal bands, the only established biomarker of MS, are detected in 0% to 20% of patients with ADEM [1]. Although there are no specific biomarkers for ADEM, serum anti-MOG-Ab and anti-aquaporin 4 antibody (anti-AQP4-Ab) tests are useful in certain cases. Anti-MOG-Ab was detected in approximately 33% to 66% of pediatric ADEM cases and 96% of ADEM cases that developed a relapsing form of demyelination [1]. Anti-AQP4-Abs are specific for NMOSD, a major type of ADEM that mimics demyelinating disorders. A case-control study showed 91% sensitivity and 100% specificity for anti-AQP4-Abs in NMOSD [38].
4. Electroencephalography
Electroencephalography (EEG) is often performed in pediatric ADEM because of seizures or altered consciousness at presentation. The most common abnormal finding is non-specific, diffuse slowing in 80% to 88% of patients [39-41]. Focal epileptiform discharges are identified in 15% to 25% of patients [39-41]. It seems that there are no significant associations between EEG findings and clinical features, or between abnormal EEG findings and epilepsy development [1].
5. ADEM subtypes
Based on the relapse and time course of ADEM, the IPMSSG has proposed an operational definition for ADEM and its variants [5]. Although debated, ADEM subtypes include ADEM, MDEM, ADEM-optic neuritis (ADEM-ON), and exceptionally acute hemorrhagic encephalomyelitis (AHEM) (Table 4). Monophasic ADEM refers to a single ADEM episode with no further demyelinating events or new MRI findings. If a relapse occurs within the first 3 months of the initial episode, it is still considered the same episode. MDEM indicates two or more episodes of ADEM separated by at least 3 months. ADEM-ON is monophasic or MDEM with one or more recurrent episodes of optic neuritis. AHEM is a severe, fatal form of ADEM with rapid deterioration and poor outcomes, and it is associated with multifocal hemorrhages and necrosis in addition to typical demyelinating lesions. Death can occur within 24 hours of onset due to severe brain edema or herniation. These diagnoses are not final because ADEM is a heterogeneous entity that can evolve into other types. As a result, it must be considered whether patients develop additional attacks or unusual presentations that are remote from the initial episode.
6. Diagnosis
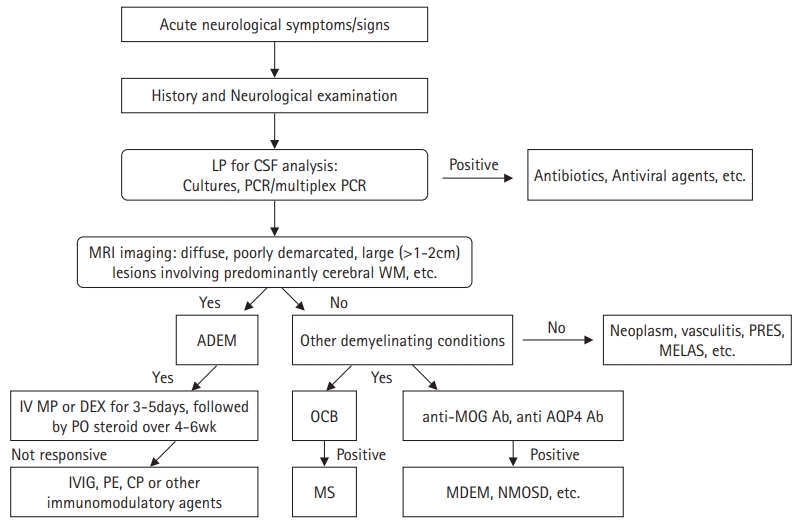
The diagnosis is still based on clinical and radiological findings and the exclusion of other conditions mimicking ADEM because there are no specific biomarkers for confirmation. The IPMSSG proposed updated diagnostic criteria for ADEM in 2013, even though the criteria are still limited in certain cases (Table 5) [5]. Taking a good history and performing a thorough physical examination are the most important first steps in the diagnosis. Children with ADEM typically present with acute encephalopathy with various multifocal neurological deficits a few days to weeks after infection or vaccination. During the acute phase, brain MRI typically diffuses bilaterally and is poorly demarcated, with large (1 to 2 cm), hyperintense lesions on T2-weighted and FLAIR sequences. Compared with adults with ADEM, MRI more often shows thalamic and basal ganglia lesions in children, and typically periventricular lesions in adults [42]. Any clinical and radiological findings occurring within 3 months of onset are considered part of the initial episode. In other words, no new clinical or MRI findings should emerge 3 months or more after the first episode of typical ADEM. When two or more episodes occur, other conditions, such as MDEM, MOG-Ab-associated disease, MS, and NMOSD, should be considered. Lumbar puncture for CSF analysis is required to rule out other ADEM-mimicking conditions, such as CNS infections.
Antibody testing, such as anti-MOG-Ab and anti-AQP4-Ab, can help define subtypes of other demyelinating disorders that are distinct from MS (Fig. 2). Demonstrating the resolution of initial MRI lesions in follow-up studies can be a supplementary method for the diagnosis of ADEM.
7. Differential diagnosis
The diagnosis of ADEM is mainly based on clinical and radiological findings owing to the lack of specific biomarkers. As a result, diagnosis often requires the exclusion of a wide variety of ADEM-mimicking conditions. Many acquired demyelination diseases and other inflammatory or non-inflammatory conditions may show similar clinical and radiological features and should be considered in terms of the differential diagnosis (Table 6).
First, potentially treatable viral or bacterial CNS infections should be ruled out. When a patient presents with movement disorders, seizures, altered consciousness, or other neuropsychiatric symptoms a few days to weeks after an infection or vaccination, autoimmune encephalitis such as anti-N-methyl-D-aspartate receptor (anti-NMDAR) encephalitis should be considered. Besides other immune-mediated demyelinating disorders, stroke-like episodes potentially indicate CNS vasculitis, systemic lupus erythematosus, moyamoya disease/syndrome, or rarely, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Bilateral thalamic involvement may be observed in children with ADEM, but it can also be noted in children with acute necrotizing encephalopathy or deep cerebral venous infarction. Bilateral basal ganglia involvement can be observed in toxic, metabolic, or genetic disorders. Bilateral cortical and subcortical lesions, predominantly in the parieto-occipital lobes, are indicative of posterior reversible encephalopathy syndrome, especially in children who are exposed to hypertension or immunosuppressive agents. If the clinical course progressively worsens, CNS tumors or genetic disorders should be considered.
Treatment
Since there are no randomized placebo-controlled studies on the treatment of ADEM, treatment protocols are mainly based on expert opinions and observational studies. Symptom-based supportive care is important because of its onset at any age or stage of illness. Supportive care can be provided together with curative treatment. Treatment with antiviral agents and antibiotics is generally accepted because ADEM mimics CNS infection.
The standard protocol is a non-specific immunosuppressive therapy including corticosteroids, intravenous immunoglobulins (IVIGs), plasma exchange (PE), and other immunomodulatory agents. Despite the lack of convincing evidence, first-line treatment usually involves a short course of high-dose corticosteroids. The most widely used protocol consists of methylprednisolone administered at 10 to 30 mg/kg/day to a maximum dose of 1 g/day or dexamethasone administered at 1 mg/kg/day, followed by oral prednisone at 1 mg/kg/day and tapered over 4 to 6 weeks [12,43,44]. In cases where corticosteroids yield poor results, IVIGs can be administered as an alternative treatment, which is administered at a total dose of 2 g/kg administered either as a single dose or divided doses over 2 to 5 days [1,12,45]. Another alternative is PE, which is usually used in children who fail to respond to corticosteroids or IVIGs (Fig. 2). PE usually involves 3 to 6 cycles with widely different protocols, and the clinical response is usually obvious after 2 to 3 exchanges [46,47]. Cyclophosphamide can be used in patients in whom conventional treatment fails, and other disease-modifying drugs such as azathioprine, mycophenolate, and rituximab can be favorable in children with recurrent MOG-Ab-positive demyelinating syndromes such as MDEM and ADEM-ON. However, clinical evidence is limited for children with ADEM [1,48].
Prognosis
The long-term overall outcome of ADEM is usually favorable because most children with ADEM respond to conventional treatment. Full recovery with normal neurological examination occurs in approximately 50% to 80% of patients, and minor neurological deficits, such as clumsiness and mild hemiparesis, are reported in 20% to 30% of patients [1,4,12]. Long-term cognitive and behavioral problems such as lower intelligence quotient, difficulties in executive function, memory impairment, and attention deficits are reported in up to 56% of patients [1,4,9,12]. According to a prior study, 22% of patients with ADEM had seizures during the acute presentation and 16.2% developed epilepsy afterward. Post-ADEM epilepsy was more frequently observed in children with relapsing disease than in those with monophasic disease and in MOG-Ab-positive children than in MOG-Ab-negative cases [49]. Although debate continues whether relapsing ADEM, called MDEM, is MOGAD or possibly NMOSD, the relapsing form has been reported in less than 10% of children with ADEM [6]. Almost a quarter of hospitalized patients with ADEM were in a critical condition and required admission to the PICU, with a mortality rate of 1% to 3% [4,6,34].
Future perspective
ADEM is an inflammatory demyelinating disease of the CNS that mostly affects children after viral or bacterial infections, or rarely after various vaccinations. The exact pathogenesis of ADEM remains unclear, although non-specific self-sensitization of reactive T cells against myelin proteins, such as MBP and myelin oligodendrocyte protein, and molecular mimicry of infectious agents are currently considered the predominant pathogenic mechanisms. Despite advances in MOG-Ab and other neurobiological clues, the diagnosis is still based on clinical and radiological findings and the exclusion of other conditions mimicking ADEM. Further studies are needed to identify any biological clues that can define the subtypes of ADEM and distinguish ADEM from other types of ADEM that mimic demyelinating diseases. Since conclusive data on the optimal treatment of ADEM and its outcomes are still lacking, well-designed collaborative multicenter studies are required to achieve the best results and minimize neurological sequelae.