A Novel Skeletal Issue in Neurodevelopmental Disorders: A Case Report of a 4-Year-Old Boy with a GRIN2B Mutation and Sacroiliitis
Article information

Glutamate ionotropic receptor N-methyl-D-aspartate type subunit 2B (GRIN2B) gene encodes GluN2B, a subunit of the N-methyl-D-aspartate (NMDA) receptor, which is closely associated with human brain development [1]. GRIN2B-related neurodevelopmental disorder presents as developmental delay or intellectual disability with other neurologic phenotypes, such as abnormal muscle tone, epilepsy, and autism spectrum disorder [2]. Early-onset findings include microcephaly, cortical malformation, and severe epileptic encephalopathy [3].
Pediatric neurologists use various diagnostic tools to identify nervous system abnormalities in children with developmental delay. For example, in a patient with movement abnormalities and developmental delay, it is essential to determine whether the condition is caused by a genetic disorder, a metabolic disorder, or a degenerative neuromuscular disorder.
The comorbidity of simultaneous developmental delay and musculoskeletal symptoms could be considered as one of the phenotypes of an underlying etiology in pediatric patients without obvious trauma or infection. However, musculoskeletal symptoms have rarely been reported in children with mutations in GRIN2B, which have been reported to be accompanied by developmental delay [4,5]. Here, we present a case of gait disturbance caused by sacroiliitis in a boy who presented with developmental delay and was found to harbor a pathogenic variant of GRIN2B upon genetic screening.
The patient was a 4-year-old boy presenting with weakness in the lower extremities with gait disturbance, who visited our clinic with no specific perinatal history. He had difficulties in feeding and had frequently experienced diarrhea since he was 3 months old. He showed normal developmental milestones until 6 months, but could not roll and sit by himself until he was 1 year old. He was able to walk independently and speak one word after 3 years of age. At the age of 4 years, he could not walk and demonstrated poor balance, swaying violently from side to side while walking or standing; he could only sit or had to be continuously embraced. He did not have any seizure-related symptoms.
His height, weight, and head circumference were all below the third percentile compared to normal children of the same age. He was unable to speak a sentence including more than two words and tried to communicate using incomprehensible sounds. He could not move his legs voluntarily, and the motor power of both legs was grade 3. The deep tendon reflexes of both knees and pulsation of the dorsalis pedis were normal. In serologic tests, inflammatory markers such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) were mildly elevated, along with leukocytosis. The initial tests for creatine kinase and its MB isoenzyme, aspartate aminotransferase, alanine aminotransferase, and lactate dehydrogenase were within normal limits. His thyroid function test, including thyroid-stimulating hormone and free thyroxine levels, showed normal results. He had a normal male karyotype (46, XY). The urine amino acid and serum organic acid profiles were within normal limits.
The chest cavity showed pectus excavatum, and an echocardiogram revealed idiopathic pulmonary hypertension; however, no evidence of thromboembolism was noted on chest computed tomography. Mild cerebral atrophy was observed on brain magnetic resonance imaging (MRI). Increased signal intensities of both sacroiliac joints and facet joints of the lumbar spine in T2-sagittal images were noted on spine MRI by our radiologist. A focal T2 hyperintense lesion with enhancement at the subcutaneous fat layer of the lower back (L3 and L4 level) was observed, which was not correlated with the clinical symptoms (Fig. 1).
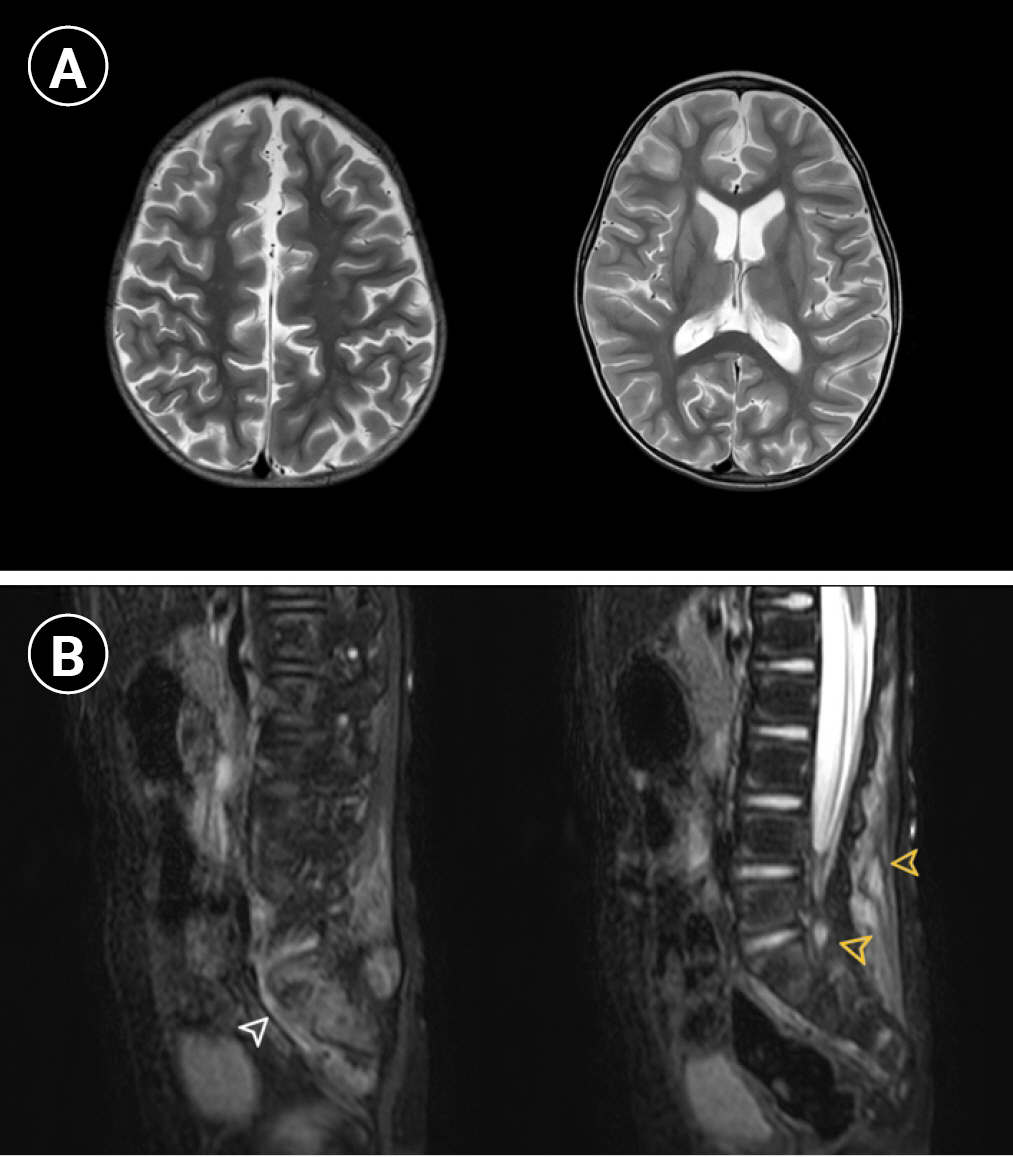
Brain and spine magnetic resonance imaging (MRI) of the patient. (A) Bilateral cerebral hemispheres show age-inappropriate diffuse atrophic changes on axial T2-weighted images. (B) Sagittal fat-suppressed T2-weighted MRI shows hyperintensity at both sacroiliac joints (white arrowhead on the left) and enhanced focal lesions in the paraspinal soft tissue and facet joints of the lower back (yellow arrowheads on the right).
We performed direct sequencing using the GC Genome diagnostic exome sequencing panel, and two heterozygous likely pathogenic variants in PLA2G6 and GRIN2B were detected. In the trio test, PLA2G6 (heterozygote missense, c.1634A>G, autosomal recessive) was detected only in the father, and GRIN2B (NM_000834.4: c.2678_2680del, p.Asn893del, autosomal dominant) was not detected in either parent; thus, the GRIN2B mutation was considered to have occurred de novo in our patient (Fig. 2). We concluded that the clinically correlated genetic pathogenic variant for this patient was GRIN2B, which is known to present various phenotypes of neurodevelopmental disorders.
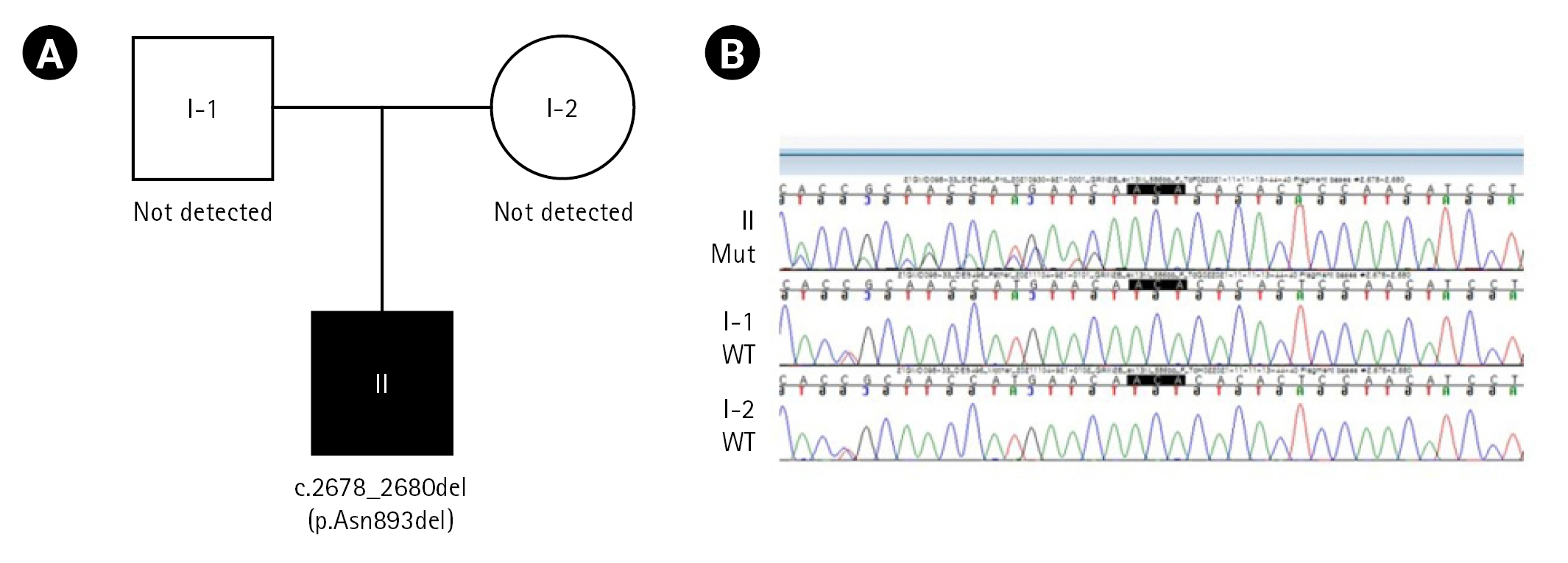
The pedigree and genetic analysis results for the family. (A) Pedigree diagram of the family. (B) Sanger sequencing analysis showing the c.2678_2680del (p.Asn893del) glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) variant in the proband.
The patient was prescribed prednisolone (10 mg daily) to attenuate joint inflammation. After 1 month, he was able to move his legs, and the serum levels of inflammatory markers (ESR, CRP) decreased to the normal range. Therefore, we decided to reduce the prednisolone dose from 10 to 5 mg per day. After initiation of the reduced dose, the patient has been able to walk without support and attempted to form sentences using more than two words after 1 month of treatment, without experiencing any adverse effects.
GRIN2B-related phenotypes with mild to severe neurodevelopmental disorders have been reported, although their phenotypes present with a wide spectrum of variation depending on genetic factors [6,7]. However, to our best knowledge, sacroiliitis associated with a GRIN2B mutation has very little support in the literature, without any specific case reports. Sacroiliitis in pediatric patients has been reported in a few cases of disorders involving immune-related mechanisms, such as juvenile spondyloarthropathies or MEFV mutations [8]. GRIN2B is mainly involved in human brain development, not the immune response. However, Canet-Pons et al. [9] reported that GRIN2B was related to the dysregulation of intracellular cholesterol transport in macrophages based on an animal experiment, and George et al. [10] reported that GRIN2B was expressed on the surface of cells in gastric cancer patients with Helicobacter pylori infections. These findings indicate that GRIN2B may have some relationship to the immune response. This report describes the first pediatric case of sacroiliitis in a patient with a GRIN2B mutation. Our report shows the possibility of that skeletal issues may co-exist with an apparent neurodevelopmental abnormality associated with the GRIN2B mutation. A GRIN2B mutation should not be excluded in children with developmental delay and gait disturbance, and we suggest a possible correlation between neurodevelopmental etiologies and other skeletal conditions.
This case report was approved by the Institutional Review Board of the Gyeongsang National University Changwon Hospital (IRB No: 2022-04-033), and written informed consent was obtained from the patient's parents.
Notes
No potential conflict of interest relevant to this article was reported. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author contribution
Conceptualization: SL. Data curation: SL, JIM, HJP, and SML. Formal analysis: SL. Methodology: SL. Project administration: SL. Visualization: SL, JIM, HJP, and SML. Writing-original draft: SL. Writing-review & editing: SL.
Acknowledgements
We thank the patient's family for participating in this study.