A Case of Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease with Acute Bilateral Total Blindness
Article information

Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD) is a demyelinating disease of the central nervous system. MOG is a glycoprotein located on the surface of oligodendrocytes that acts as a cellular adhesive molecule in the central nervous system. The role of MOG has not been fully elucidated, but it is known to regulate microtubule stability and modulate myelin-immune interactions by mediating the complement cascade [1]. MOGAD clinically presents with optic neuritis, transverse myelitis (TM), or rarely, acute disseminated encephalomyelitis (ADEM), depending on the location of the lesion. The clinical manifestations of MOGAD differ by age. Younger children mostly present with an ADEM phenotype. In contrast, optic neuritis is commonly observed in older children and adults [2]. Since the symptoms correlate with the location of the demyelinating lesion, it is often difficult to distinguish between multiple sclerosis (MS), aquaporin-4 antibody-associated neuromyelitis optica spectrum disorder (AQP4-NMOSD), and monophasic acquired demyelinating syndrome (ADS). These demyelinating diseases were traditionally classified according to recurrence. Since the early 2000s, advances in diagnosis using antibody assays have led to the recognition of MOGAD as a new disease entity distinct from other demyelinating diseases [3]. Herein, we present a case of MOGAD with complete remission following a combination of plasmapheresis, intravenous immunoglobulin (IVIG), and steroids. This study was approved by the Institutional Review Board of the Gangnam Severance Hospital, Yonsei University College of Medicine (3-2017-0168). The requirement for informed consent for this retrospective study was waived by the board.
A 12-year-old girl complained of progressive visual loss for 3 days and became completely blind. She had a history of an upper respiratory tract infection a week prior to symptom onset. The visual defect started in the right eye and progressed to the left eye, eventually affecting both eyes. Eye examination revealed decimal visual acuity of 0.1/0.3 (right/left). A fundus examination with pupil dilation revealed a swollen optic disc in both eyes but no disc redness, retinal hemorrhage, or changes in the retinal vessels. NMOSD, ADEM, MS, and other central nervous system inflammatory diseases were considered in the differential diagnosis. The patient was treated with methylprednisolone (1 g/day) for 5 days. However, her vision did not improve, and she was referred to a tertiary hospital.
On admission, the patient’s visual acuity had deteriorated to hand motion in the right eye and a finger count of 10 cm in the left eye. Automated perimetry revealed complete depression of the visual field in both eyes. Blood tests and cerebrospinal fluid (CSF) analyses were performed, and the results were normal. The C-reactive protein level and erythrocyte sedimentation rate were normal. CSF analysis revealed normal protein levels with a slight increase in the white blood cell count and negative oligoclonal bands. Brain magnetic resonance imaging (MRI) revealed bilateral optic neuritis, including the optic nerves, optic chiasm, and focal abnormalities in the subcortical white matter and cortical gray matter of both insula (Fig. 1A and B). There was no evidence of demyelination of the spinal cord. Six cycles of plasmapheresis were initiated, followed by IVIG at 2 g/kg for 2days.
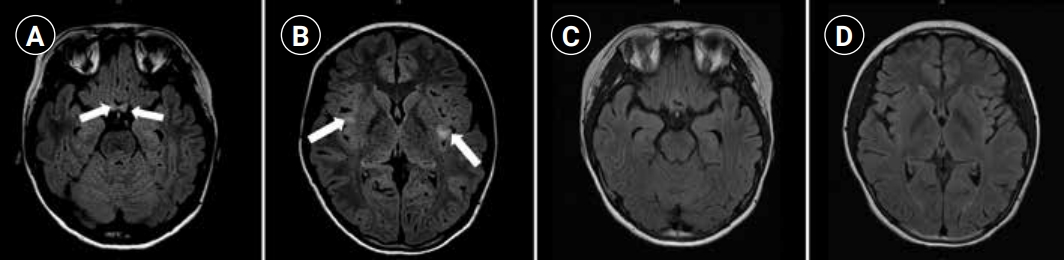
Magnetic resonance imaging (MRI) findings. The initial axial Fluid attenuated inversion recovery images revealed bilateral optic neuritis (arrows, A) and focal abnormalities in the subcortical white matter and cortical grey matter in insula (arrows, B). Follow-up MRIs revealed resolving process of bilateral optic neuritis and resolved state of both insula lesions (C, D).
During admission, serum MOG antibody (MOG immunoglobulin G) was tested using a fluorescence-activated cell sorting live cell assay. It tested positive, with a positive cell ratio of 0.422 (negative, ≤0.141; borderline, >0.141 or ≤0.254; positive, >0.254). Serum anti-aquaporin 4 antibody was negative. We were able to confirm her diagnosis as MOGAD. Ophthalmic examinations were performed on consecutive days after each cycle of plasmapheresis. The patient’s visual acuity and visual field improved progressively. After six cycles of plasmapheresis, the MOG antibody level test revealed a negative conversion. At discharge, the patient’s visual acuity improved to 0.04/0.02 (right/left), and the visual field also improved to 71%/31% (right/left). She was discharged with a prescription of oral prednisolone at 1 mg/kg/day and monthly administration of IVIG (1 g/kg) until full recovery of visual acuity.
One month after discharge, the patient visited our hospital for her monthly IVIG treatment and follow-up MRI. The brain MRI demonstrated resolution of the bilateral optic neuritis, including the optic nerves and optic chiasm, and there were no newly developed lesions on follow-up MRI (Fig. 1C and D). There was also no evidence of demyelination in the spinal cord on spinal MRI. An ophthalmic examination revealed a visual acuity of 0.08/0.06 (right/left) and a visual field of 65%/71% (right/left). Owing to the side effects of oral prednisolone, such as iatrogenic Cushing syndrome, we prescribed deflazacort to the patient instead of prednisolone and gradually tapered off. She regularly had outpatient department visits for routine check-ups. Three months after treatment, an examination revealed further improvement in visual acuity (0.9/0.8, right/left). Three cycles of monthly IVIG were administered, and the patient’s visual acuity almost returned to the pre-morbid level, with a visual field of 94%/97% (right/left) (Fig. 2). There was no recurrence during the 4-month observation period, and the patient remained seronegative for the MOG antibody.
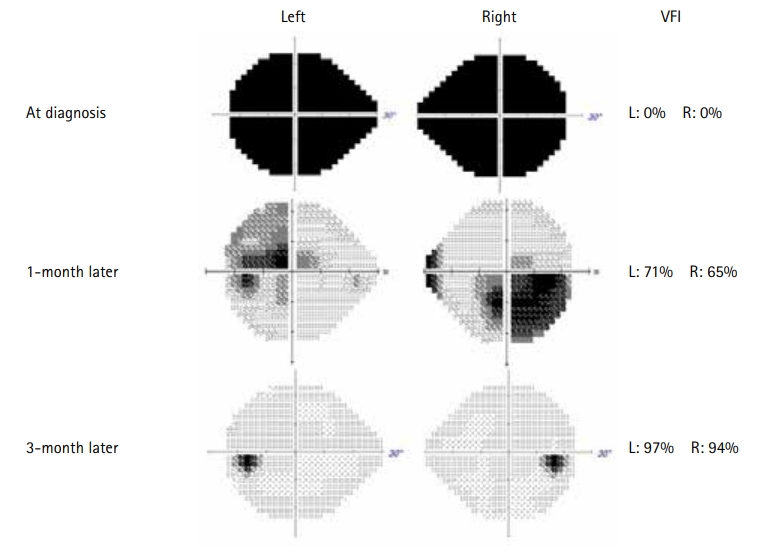
Improvement of visual field index (VFI) over time after treatment.
Between 0.5 and 1.66 per 100,000 children have ADS. When children first present with ADS, the differential diagnosis is crucial because specific and timely treatment may be required [4]. MOGAD, MS, and AQP4-NMOSD are the common diagnostic possibilities. One-fifth of children with ADS are diagnosed with MS, and MOG-Ab is found in approximately 30% of children with ADS [3]. Optic neuritis and TM are the frequent symptoms in all three diseases, whereas ADEM is present in MOGAD and AQP4-NMOSD, but rarely in MS. Whereas monocular optic neuritis is common in MS, bilateral eyes are affected in MOGAD and AQP4-NMOSD. The diagnosis should be confirmed based on laboratory findings, especially an antibody assay [3].
Management of an acute demyelinating event is key to reducing inflammation and promoting recovery without sequelae. The first-line treatment involves intravenous steroids (1–2 g/day for 5–7 days). If the patient is resistant to steroids, plasma exchange or IVIG is used as second-line treatment. Considering the severity and risk of relapse, chronic immunotherapy may be needed to prevent relapse. Rituximab, azathioprine, tacrolimus, mycophenolate mofetil, and methotrexate are commonly used [5]. In MS, initial disease-modifying treatment is recommended for better control of clinical and radiological disease activity owing to a higher relapse rate. However, MOGAD tends to be monophasic, and the disease-modifying treatment used for MS is not suitable [6].
The clinical course of MOGAD may be monophasic or relapsing [1]. Younger children or those with the ADEM phenotype tend to have monophasic experiences, and persistent seropositive MOG antibodies may be associated with a higher relapse rate. In particular, the relapse rate is about 50% in MOG-optic neuritis (ON), and regular follow-up is needed, including ophthalmic examinations, neurologic examinations, and MOG antibody level tests [7]. The severity of relapse is variable, but most children recover quickly. Almost complete remission is usually seen [2]. The severity is also age-dependent. Symptomatic brain involvement is more likely to occur in younger children than in older children [8].
In conclusion, we present a case of MOGAD manifesting as optic neuritis that was treated with steroids, plasmapheresis, and IVIG. The patient recovered, with MOG antibody seronegativity and no relapse. A systematic review of pediatric MOGAD included 61 studies, all but one of which used corticosteroids as acute-phase treatment. However, IVIG was used in 21% (128/621) of patients and plasmapheresis was only performed in 4% (26/621) [9]. MOGAD is commonly well treated with intravenous steroids, but cases in which the patient is refractory to steroids or presents with severe symptoms such as total blindness could be managed with plasmapheresis and IVIG for a better prognosis, as observed in our case.
Notes
Young-Mock Lee is an editorial board member of the journal, but he was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: JK, JHN, and YML. Data curation: JK, JHN, JCB, JSK, and YML. Methodology: JK, JHN, and YML. Visualization: JK, JHN, and YML. Writing-original draft: JK. Writing-review & editing: JK, JHN, HL, and YML.
Acknowledgements
The authors are grateful to all staff members, doctors, and statistical consultants who were involved in this study.