Neonates are vulnerable to epileptic seizures. Eighty percent of neonatal seizures occur in the first 1 or 2 days of life. The most common cause of neonatal seizures is hypoxic ischemic encephalopathy. Brain malformations can also be an important cause of seizures. Herein, we present a case of an infant who experienced severe myoclonic seizures with little response to anti-seizure medications from the first day of life. The infant had refractory myoclonic seizures associated with pontocerebellar hypoplasia (PCH) and a mutation in transfer ribonucleic acid splicing endonuclease 54 (TSEN54). This case was reviewed and approved by the Institutional Review Board of Keimyung University Dongsan Hospital (IRB No. 2022-04-017). The requirement for informed consent was waived by the board.
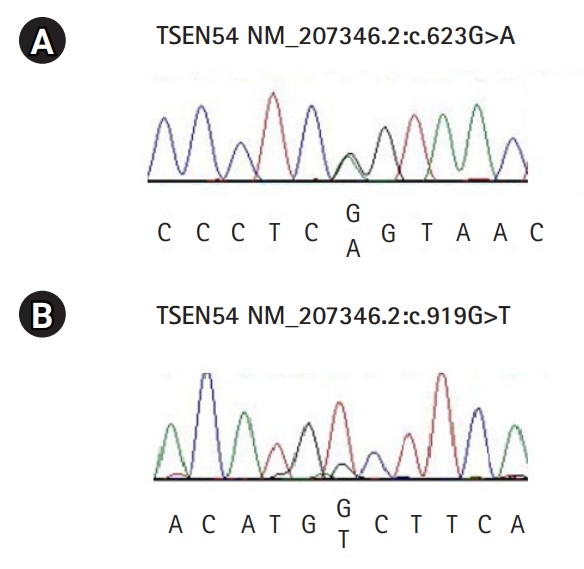
A female infant was born at 38 weeks of gestation by cesarean section due to breech presentation at a local hospital. She had a birth weight of 2,840 g (25th to 50th percentile), length of 45 cm (10th to 25th percentile), and head circumference of 31 cm (<10th percentile). Although her Apgar scores were 7 and 9 at 1 and 5 minutes, respectively, the baby suffered from severe seizures and respiratory distress from the first day of life. Therefore, she was transferred to a university hospital. A neurological examination revealed generalized myoclonic seizures with respiratory distress, hypertonia of the extremities, joint stiffness of both elbows, and increased deep tendon reflexes. Initial electroencephalography (EEG) showed frequent ictal EEG patterns with 6 to 7 Hz rhythmic activities beginning at P3, P4, and T6 independently. Radiologic studies (Fig. 1) showed a flat ventral pons and a small cerebellum. This baby had no specific findings in studies for metabolic disorders or genetic screening tests associated with early myoclonic seizures in infancy. To determine the genetic cause of PCH, targeted exome sequencing was performed when she was 1 month of age. Finally, two variants of the TSEN54 gene, c.919G>T and c.623G>A, were found, representing compound heterozygosity. These two variants were confirmed by Sanger sequencing (Fig. 2), and no other copy number variation was found in the diagnostic exome sequencing study. The c.919G>T mutation (NM_207346.2:c.919G>T, p.Ala307Ser, rs113994152) has been already reported as the most common TSEN54 variant found in PCH patients [1,2], and has been classified as a pathogenic variant in the ClinVar database. It is very rare in large population databases, such as gnomAD (https://gnomad.broadinstitute.org/), where it has a minor allele frequency (MAF) of 0.09%, and Korean Reference Genome Database (KRGDB; http://coda.nih.go.kr/coda/KRGDB/), where it has a MAF of 0%. This variant was also predicted to be deleterious in an in silico analysis using Sorting Intolerant From Tolerant (SIFT; https://sift.bii.a-star.edu.sg/) and Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/). Based on this evidence, the c.919G>T variant was classified as a likely pathogenic variant. The other mutation, c.623G>A (NM_207346.2:c.623G>A, Arg208Gln, rs85291542), is also a very rare variant (GnomAD, MAF=0.0008% and KRGDB, MAF=0.00%), and has been not reported in any articles or databases. An in silico prediction suggested that this variant is likely to have a splicing effect by splice site analysis, and it was identified as a disease-causing mutation using MutationTaster (http://mutationassessor.org/). According to the American College of Medical Genetics guideline, c.623G>A was classified as a variant of unknown significance. However, based on its rarity in the general population and consistent clinical findings, this variant has a high probability of being a pathogenic cause in this patient, although there is a limitation in interpretation since the patientŌĆÖs family was not tested.
After two and a half months of hospitalization, the patient had brief myoclonic seizures that persisted several times a day, with no noticeable improvement. At discharge, neurological examination revealed that hypertonia of the extremities, joint stiffness, and abnormal ankle myoclonus were still present. The baby was discharged from the hospital with assisted ventilation, gavage, and anti-seizure medications, including carbamazepine and levetiracetam.
PCH is a rare genetic neurodevelopmental disorder mostly characterized by prenatal developmental disorders and deterioration of cerebral structures [3]. Of the 13 known subtypes, PCH2 is the most common form of autosomal recessive PCH [4,5]. The age at onset in PCH2 varies from prenatal to early infancy. Most patients present with this condition in the first months of life. Patients with PCH2 have symptoms including microcephaly, abnormal muscle tone, epilepsy, and severe psychomotor retardation [6]. In our case, the patient had microcephaly, hypertonia of extremities, joint stiffness of both elbows, and generalized myoclonic seizures with respiratory distress. Both PCH2 and 4 are caused by mutations in the TSEN54 gene, and both exhibit an absence of transverse pontine fibers and underdevelopment of the cerebellar folia. Early-onset infantile PCH has numerous causes, including chromosomal disorders (e.g., trisomy 13, 18, or 21), metabolic disorders (e.g., a congenital disorder of glycosylation type 1a or glutaric aciduria), exposure to teratogens (e.g., alcohol, anticonvulsant therapy, or infection with cytomegalovirus), syndromic associations (e.g., Bowen-Conradi syndrome or Marden-Walker syndrome), and an emerging group of autosomal recessive single-gene disorders [7,8]. In our patient, the cerebellar hemispheres were flat and very small, whereas the cerebellar vermis was relatively conserved. In simple cerebellar atrophy, the pons volume is maintained relatively well. Joubert syndrome shows characteristic molar tooth signs. Typical PCH2 brain magnetic resonance imaging (MRI) findings include a characteristic dragonfly shape in the coronal plane [1,3,9]. Supratentorial involvement is also possible. It is reflected by variable neocortical atrophy, ventriculomegaly, and microcephaly. The MRI findings of the present case were consistent with PCH. Causative mutations in genes that encode 3 of the 4 subunits of the TSEN complex, namely TSEN54 (MIM 277470), TSEN2 (MIM 612389), and TSEN34 (MIM 612390), have been found in most cases with PCH2 [3,8]. Of these, the ŌĆ£commonŌĆØ c.919G>T/p.A307S variant, TSEN54, accounts for approximately 70% of mutated PCH alleles and more than 85% of all PCH2 cases [10]. Our case also showed the most common mutation of TSEN54. To the best of our knowledge, this is the first case report in Korea of PCH2 that was confirmed by genetic testing.