Expanding the Clinical and Genetic Spectrum of Caveolinopathy in Korea
Article information

Abstract
Purpose
Caveolinopathy is a disease caused by caveolin-3 (CAV3) mutations that shows a wide clinical spectrum, including isolated hyperCKemia and limb-girdle muscular dystrophy. While recent advances in next-generation sequencing (NGS) have enabled earlier diagnosis of this disease, it remains difficult to predict the clinical course of each patient.
Methods
This study summarizes the clinical presentations of 13 genetically confirmed caveolinopathy patients in four Korean families. Genetic diagnosis was performed using NGS technologies for probands and Sanger sequencing for the other family members.
Results
Four coding mutations were found (p.Val103_Val104del, p.Asp28Glu, p.Pro105Leu, and p.Arg27Gln), and each family showed autosomal dominant inheritance. While all 13 cases had hyperCKemia, only five of them showed some myopathic features including ankle contracture, calf hypertrophy, exercise intolerance, and muscle cramping. This high proportion of asymptomatic cases suggests both that these mutations may be associated with a mild phenotype and that caveolinopathy may be an underdiagnosed disease.
Conclusion
This study extends our understanding of caveolinopathy; in particular, the findings suggest the need to consider caveolinopathy in patients with incidental findings of creatine kinase elevation. NGS may be a useful method in the differential diagnosis of such cases.
Introduction
Caveolinopathy is a disease caused by alterations in caveolin-3 (CAV3), a muscle-specific protein encoded by CAV3 [1]. It has been designated as LGMD1C, a subtype of limb–girdle muscular dystrophy (LGMD), but its clinical presentations are highly heterogeneous, including rippling muscle disease (RMD), LGMD, long QT syndrome, distal myopathy, and isolated hyperCKemia [2]. Although the prevalence of caveolinopathy is unclear, it seems evident that it is a fairly rare disease. LGMD1C accounts for fewer than 5% of cases of LGMD, whose prevalence ranges from 1 in 14,500 to 1 in 123,000 according to ethnicity [3]. One study on an LGMD cohort in the USA reported only one CAV3-affected case out of 4,656 patients [4], and there are currently around 50 mutations of CAV3 listed in the Human Gene Mutation Database (HGMD) [5].
The progression of muscle weakness may be slow or rapid. The age of onset varies greatly from early childhood to late adulthood, and initial symptoms also differ among individuals [6-8]. In addition, the same mutations of CAV3 may result in quite different clinical features, showing evidence of intrafamilial phenotypic variability [2,9]. These diverse clinical presentations of caveolinopathy have posed a diagnostic challenge. However, in recent decades, advances in next-generation sequencing (NGS) technology have increased the diagnostic rate of caveolinopathy and further broadened its clinical spectrum. In addition to NGS, conventional diagnostic methods, such as muscle pathology and immunohistochemical staining, can still be helpful in confirming the pathogenicity of variants [10].
To our knowledge, few studies have investigated caveolinopathy in East Asian populations [11,12]. Although a couple of Korean patients have been reported previously [10,13], no caveolinopathy case series has been published to date. Considering its rarity and heterogeneity, a broad range of studies is warranted among different ethnicities to elucidate the clinical and genetic spectrum of caveolinopathy. In this study, we summarized the clinical manifestations of 13 caveolinopathy cases in four Korean families, which were diagnosed using NGS technology and reviewed through muscle pathology. We investigated whether there were any differences in clinical presentations according to the different types of mutations.
Materials and Methods
1. Subjects
The study was approved by the Institutional Review Board (IRB) of Seoul National University Hospital (IRB no. 2008-135-115), and written informed consent was obtained from all patients and/or their parents. In total, 13 patients with proven mutations were enrolled in this study. They belonged to four families, and the largest family included eight affected patients (Fig. 1). The probands visited the Neuromuscular Clinic of Seoul National University Children’s Hospital (SNUCH) with symptoms or signs suspicious of myopathy such as hyperCKemia and a tiptoeing gait, and they were all genetically diagnosed with caveolinopathy with CAV3 mutations. We found nine cases with CAV3 mutations in relatives through mutation screening in their family members. We retrospectively reviewed their medical records, including laboratory, radiographic, and pathologic findings during follow-up, and compared them within and between families according to their mutations.
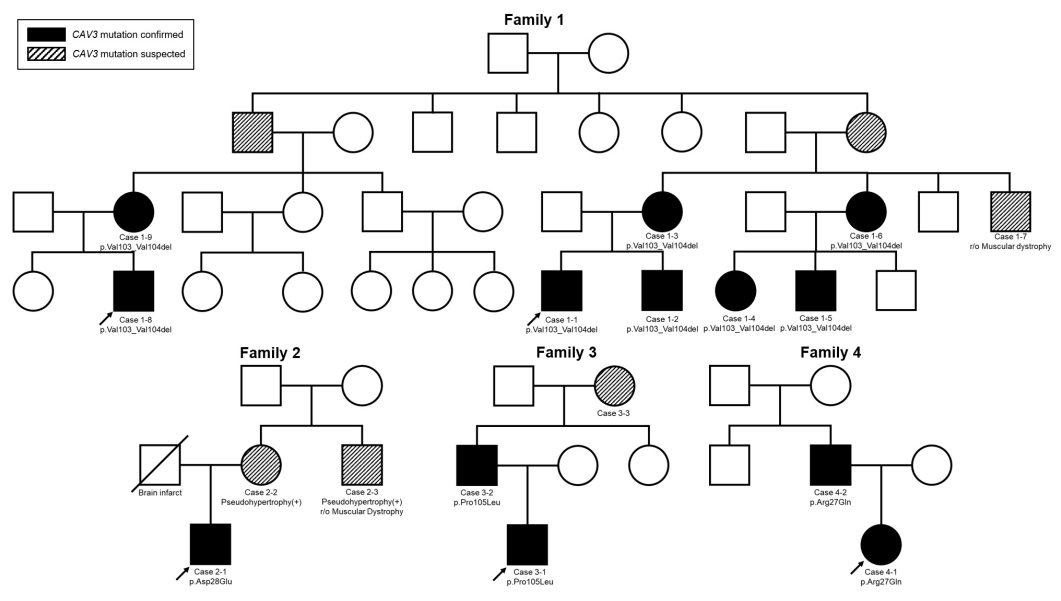
Pedigrees of the families affected by caveolinopathy. Four independent families carried different caveolin-3 (CAV3) mutations, all inherited in an autosomal dominant manner.
2. Mutation discovery
With the suspicion of isolated hyperCKemia or muscular dystrophy, we tried to find causal variants for each patient (Fig. 2). Single-gene analysis of the dystrophin (DMD) gene could not identify pathogenic variants, and the probands of each family were sequenced using different types of NGS technologies. Cases 1-1, 1-8, and 4-1 were sequenced through the LGMD panel of SNUCH. This panel is designed to include the exonic regions of 43 LGMD-related genes, using the Agilent SureSelectXT Custom kit (Agilent Technologies, Santa Clara, CA, USA). Case 2-1 was sequenced through whole-exome sequencing using the SureSelectXT2 Human All Exon v4+UTRs kit (Agilent Technologies) and HiSeq 2500 (Illumina, San Diego, CA, USA). The mutation in case 3-1 was found using a commercial NGS platform (Diagnostic Exome Sequencing, Green Cross Genome, Yongin, Korea).
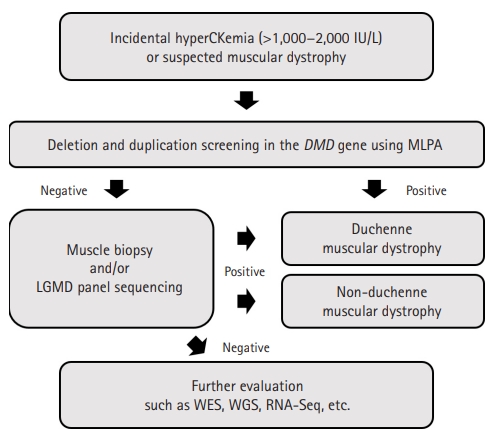
Diagnostic approach to patients with incidental hyperCKemia or suspected muscular dystrophy. In general, we first apply multiplex ligation dependent probe amplification (MLPA) of dystrophin (DMD) to the patients. If negative, we conduct muscle biopsy or limb-girdle muscular dystrophy (LGMD) gene panel sequencing, sometimes both at the same time. Further studies such as whole-exome sequencing (WES), whole-genome sequencing (WGS), and RNA sequencing (RNA-Seq) are considered if the above studies fail to find causal mutations.
The discovered variants were predicted to be “pathogenic” or “likely pathogenic” according to the guideline suggested by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology in 2015 [14]. HGMD was used to check whether the mutations had been previously reported. Public variant databases, such as the Genome Aggregation Database (gnomAD), were screened for their population allele frequencies [15]. Finally, the discovered mutations were confirmed in other relative cases using Sanger sequencing. The details of the sequencing process and variant analysis were described previously [16].
3. Immunohistochemistry
Muscle biopsy was conducted in cases 1-1, 1-8, 2-1, and 3-2. Muscle samples were taken from the quadriceps femoris or rectus femoris and frozen with isopentane cooled in liquid nitrogen. Serial frozen sections of 10 μm were stained with a set of histochemical stains. Purified mouse anti-caveolin-3 antibody (BD Transduction Laboratories, Lexington, KY, USA) was used for immunohistochemical staining.
Results
1. Clinical presentations and comparison with causal mutations
The clinical and mutational characteristics of the study samples are summarized in Table 1. Each sample with a CAV3 mutation had hyperCKemia regardless of their age, sex, and symptoms. Four CAV3 mutations were detected in four different families using NGS technologies, inherited in an autosomal dominant manner. All the mutations were predicted to be pathogenic or likely pathogenic and previously reported in the HGMD. In addition, none of them was listed in the public variant database, gnomAD.

Clinical and genetic information of the study subjects
1) Family 1
All the cases genotyped in family 1 shared the same mutation, namely a heterozygous in-frame deletion of CAV3 c.307_312delGTGGTG (p.Val103_Val104del). Case 1-1 visited the clinic after an incidental finding of elevated creatine kinase (CK) concentration (564 to 1,378 IU/L; reference range, 20 to 270 IU/L) at 13 years of age. He was previously healthy and achieved normal motor developmental milestones except for being a slow runner during early childhood. While showing Achilles tendon tightness and calf hypertrophy on physical examination, he had normal motor power until the last follow-up (at 17 years old).
Case 1-2 was a younger brother of case 1-1. Likewise, while he had hyperCKemia, ankle contractures, and muscle cramping during exercise at 8 years of age, calf hypertrophy was not observed, and his motor performance was almost normal. Case 1-5, a maternal cousin of case 1-1, had hyperCKemia and muscle cramping without ankle contracture at 9 years of age. In addition, a 39-year-old maternal uncle (case 1-7) had been clinically diagnosed with muscular dystrophy and showed tiptoeing gait, although symptom progression was not definite. However, his DNA sample was not available and the presence of a mutation could not be confirmed. Other female family members (cases 1-3, 1-4, and 1-6) had no symptoms or signs, except for elevated CK levels.
Case 1-8 visited the clinic with an isolated hyperCKemia at 7 months of age, and calf hypertrophy and ankle contracture developed during follow-up. Despite showing normal development and tolerable motor performance, he started to have mild and intermittent exercise intolerance at the age of 16 years. Additionally, he started to show seizures at age 15 years, and there were four episodes of sudden collapse with loss of consciousness followed by generalized tonic-clonic seizures. He was treated with an antiepileptic medication (divalproex sodium) with a clinical diagnosis of idiopathic generalized epilepsy. Electroencephalography and 24-hour Holter monitoring showed no definite abnormal findings, and no further seizures were detected since the start of treatment. His mother, case 1-9, had isolated hyperCKemia without any motor symptoms or signs until the fourth decade.
2) Family 2
Case 2-1 had a missense mutation, CAV3 c.84C>A (p.Asp28Glu). He was the only mutation-proven patient with definite exercise intolerance and tiptoeing gait in this study. He started to show tiptoeing gait from 4 to 5 years old, and visited the clinic for a further evaluation at 16 years old. He had hyperCKemia, calf hypertrophy, exercise intolerance, and muscle cramping at that time, but his motor power was not definitely impaired. His mother (case 2-2) was shown to have isolated hyperCKemia (694 IU/L), and his maternal uncle (case 2-3), whose mutational status was unknown, also had similar muscle symptoms and was discharged early from military service due to the symptoms. Additionally, case 2-1 had major depressive disorder with suicidal ideation and was treated with antidepressants.
3) Family 3
The members of family 3 shared a CAV3 c.314C>T (p.Pro105Leu) mutation. Case 3-1 had a history of birth asphyxia and suspicious muscle hypotonia at birth and remained in a neonatal intensive care unit for 1 month. At that time, CK elevation was incidentally found and his father brought him to the clinic for further evaluation at 2 months of age. He had no abnormal symptoms or signs and started to control his head, and he showed a social smile at the initial visit. He showed normal development until the last follow-up (at 30 months of age), walking well without support. His father and grandmother (cases 3-2 and 3-3) had no definite exercise intolerance or other muscle symptoms, but their CK levels were elevated (1,713 to 1,847 and 1,386 IU/L, respectively).
4) Family 4
Case 4-1 inherited a missense variant, CAV3 c.80G>A (p.Arg27Gln), from Case 4-2. Case 4-1 visited the clinic due to hyperCKemia at 4 years of age (320 to 527 IU/L), which was incidentally found during treatment for Kawasaki disease at an outside hospital. CK levels were measured in both parents, and her father (case 4-2, 36 years old) was also shown to have CK elevation (813 IU/L). However, neither case 4-1 nor 4-2 had any muscle symptoms or signs at the time of the study.
We also performed electrocardiography (cases 1-1, 1-8, 2-1, and 3-2) and echocardiography (cases 1-1, 1-5, 1-8, and 2-1) for cardiac evaluation. The results showed no abnormal findings and normal heart function, indicating no definite cardiac muscle involvement at that time. Four patients (cases 1-1, 1-2, 1-5, and 2-1) underwent electromyography and nerve conduction studies, and the results were compatible with myopathy, showing increased insertional activity, short duration, reduced-to-complete interference pattern, and an early recruitment pattern.
2. Muscle pathology
We conducted muscle biopsy for four cases (cases 1-1, 1-8, 2-1, and 3-2). All findings were consistent with muscular dystrophy, showing several myopathic changes such as size variation, degeneration and regeneration of myofibers, and endomysial/perimysial fibrosis with fatty changes. Although case 3-2 was 39 years old at the time of biopsy, the oldest among them, his muscle pathology was the least severe, showing minimal size variation of myofibers and minimal endomysial fibrosis.
While immunohistochemical staining for dystrophin 1, 2, and 3 showed no definite abnormalities, the signal activities of dysferlin were weakly positive and negative in cases 1-1 and 2-1, respectively. Therefore, those cases were initially suspected of having dysferlinopathy before genetic diagnosis. We also performed immunohistochemical staining using antibody to caveolin, which revealed a definitively low expression compared with normal controls (Fig. 3).

Immunohistochemical staining of caveolin-3 in muscle biopsies. Sarcolemmal labeling of caveolin-3 (1:1,000 dilution) showed reduced staining in all three unrelated patients (Case 1-1, Case 2-1, and Case 3-2) compared with normal controls.
Discussion
This study reported 13 caveolinopathy cases in four Korean families. We found four CAV3 mutations and summarized their clinical presentations in association with their causal mutations. To our knowledge, this is one of the largest clinical studies on caveolinopathy, especially in an East Asian population. Although all mutations were previously reported and listed in HGMD, the clinical features differed from each other even when the same mutations were involved.
Except for case 2-1, who visited for a further evaluation of tiptoeing gait, each patient visited the clinic with a chief complaint of hyperCKemia, and muscle symptoms were mostly mild or absent during follow-up. Although caveolinopathy patients can have diverse clinical presentations, including LGMD, RMD, and long QT syndrome [2], the mutations in our cases (p.Val103_Val104del, p.Pro105Leu, and p.Arg27Gln) might be more associated with mild types of caveolinopathy. The p.Asp28Glu mutation of family 2 was previously reported in a large German family with RMD and LGMD [9]. That study showed that the affected patients did not have muscle weakness, except for ankle dorsiflexion, until their late 30s. Even when the disease progressed to muscle weakness, the study patients had muscle power of no less than grade 4 until the sixth decade of life. The p.Pro105Leu missense mutation of family 3 was also previously found in two Italian families with LGMD [17]. In that study, while disease onset usually occurred at 5 years of age, patients showed normal motor development, mild-to-moderate proximal muscle weakness, and slow disease progression. Likewise, case 3-3, the grandmother of case 3-1, had no definite exercise intolerance at the time of the study, and the muscle biopsy of case 3-2 showed relatively favorable pathology compared with that of the other mutations. Regarding the p.Arg27Gln mutation, there is one case report in a woman who had no neuromuscular symptoms until the age of 70 years [18].
Case 2-1, with the p.Asp28Glu mutation, had more severe symptoms than the other cases in this study; we assume that the difference in mutation position resulted in different clinical features. However, it is early to draw clear conclusions because of the small sample size and short follow-up period. In addition, considering the intrafamilial phenotypic variability of caveolinopathy [2,9], it is difficult to predict the clinical course of each patient according to the type of causal mutation.
Some patients suffered from neuropsychiatric disorders: case 1-8 with epilepsy and case 2-1 with depression. Although the relationship between CAV3 and neuropsychiatric disorders has not been well established, one recent study found an association between CAV3 variants and epilepsy [19]. This group also reported that CAV3 mutations could alter the current of hyperpolarization-activated cyclic nucleotide-gated channels and may develop cardiac arrhythmias [20]. However, given the limited available data and the absence of additional genetic or molecular studies conducted for these patients, more evidence is needed to support any association between CAV3 and neuropsychiatric disorders.
Interestingly, in this study, the cases with myopathic features, such as ankle contracture, muscle cramping, exercise intolerance, and tiptoeing gait, were all males, and we speculate that male caveolinopathy patients might have earlier onset and more severe symptoms compared with females. The p.Val103_Val104del mutation has also been reported in a male patient with RMD who showed muscle weakness and atrophy with symptom onset at 17 to 18 years of age [13]. While his mother and elder sister had the same mutations, they had no definite muscle weakness or atrophy. This pattern agrees with our findings showing more severe clinical features in male patients. However, further studies are required to confirm this difference by sex, which might originate from other genetic or environmental factors such as differences in the level of physical activity.
Two patients, cases 1-1 and 2-1, were initially suspected of having dysferlinopathy because of decreased signal activities of dysferlin on immunohistochemical staining. One study noted that two sarcolemmal proteins, dysferlin and CAV3, interact with each other in skeletal muscle, and CAV3 mutations can result in the deterioration of dysferlin [21]. In contrast, dysferlin impairment may also result in decreased CAV3 expression [22].
We previously reported the p.Val103_Val104del mutation of cases 1-8 and suggested its ethnic specificity and the possibility of asymptomatic or minimally affected carriers in the Korean population [10]. Since none of the four mutations in this study were listed in either gnomAD or our in-house database of around 1,000 Korean genomes, further sequencing is required to reveal the allele frequencies of these rare variants.
In conclusion, this study showed the clinical spectrum of caveolinopathy in patients with four different mutations. Because all the patients in this study had mild myopathic features, caveolinopathy needs to be considered in patients with no or mild muscle symptoms with hyperCKemia. However, considering the phenotypic variability, each patient needs to be checked regularly regardless of the presence and severity of clinical symptoms. Other genetic and environmental factors may determine the clinical presentation of caveolinopathy in addition to CAV3 mutations, and further research is needed to elucidate the genotype–phenotype correlation in more detail and predict the clinical course of each patient.
Notes
Ki Joong Kim, Jong-Hee Chae and Anna Cho are the editorial board members of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: JHC and AC. Data curation: SL, SYK, and BCL. Formal analysis: SL. Project administration: KJK and JHC. Visualization: SL. Writing-original draft: SL. Writing-review & editing: AC.
Acknowledgements
This research was supported by research funding (2021-ER0701-01) from the Korea Disease Control and Prevention Agency.