Effects of Salbutamol in Collagen like Tail Subunit of Asymmetric Acetylcholinesterase-Related Congenital Myasthenic Syndrome: A First Korean Case
Article information

The congenital myasthenic syndrome (CMS) is a heterogeneous group of disorders of the neuromuscular junction [1]. Suspecting and performing a molecular genetic testing is important in CMS considering that the symptoms in this group of disorders are similar, but treatments vary. The syndrome can be classified as presynaptic, synaptic basal lamina associated, and postsynaptic according to the location of the defects and genetic diagnosis [2]. Administration of acetylcholinesterase (AChE) is often the treatment of choice, but caution is required because AChE can aggravate the symptoms in collagen like tail subunit of asymmetric acetylcholinesterase (COLQ)-related CMS (Table 1) [3-5]. Here, we report a first Korean female patient who was diagnosed with COLQ-related CMS with muscle disease gene panel.
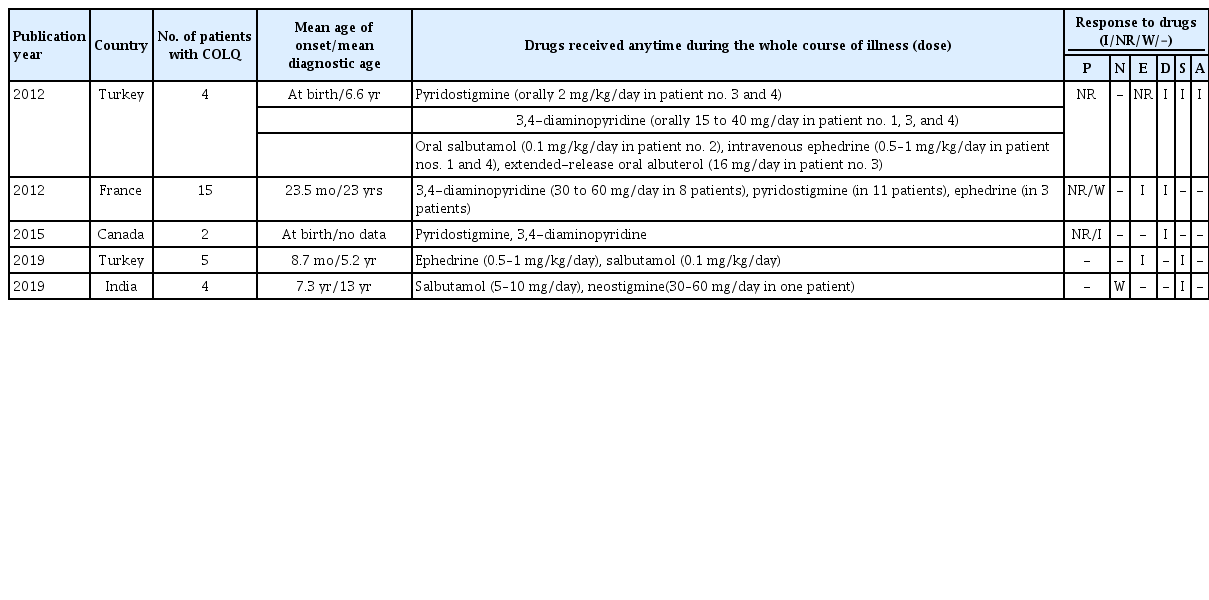
Summary of results of a few previous COLQ-mutant congenital myasthenic syndrome studies
A 2-year-old girl was transferred to the pediatric intensive care unit (PICU) of our hospital due to respiratory failure. This was her seventh admission to the PICU due to respiratory difficulty. At the age of 10 days, she developed paroxysmal cyanosis, poor oral intake, and decreased activity and was admitted to the other hospital for the first time due to respiratory failure without pathogen. At the age of 2, 4, 6, and 8 months, she was readmitted due to respiratory failure without pathogen. Only at the age of 11 months, she was admitted to the PICU due to respiratory syncytial virus infection.
On admission, she looked dysmorphic with myopathic face, high arched palate, midfacial hypoplasia and micrognathia. On physical examination, she had low body weight (5.6 kg, <3rd percentile), normal height (70 cm, 77th percentile), and normal head circumference (41.5 cm, 24th percentile). Physical examination also showed shallow breathing, neck flexor weakness, and generalized proximal dominant motor weakness in both upper and lower extremity, though there were no scoliosis, apparent muscle atrophy, and contractures. However, on neurologic examination, she had a normal deep tendon reflex, and Babinski reflex was not observed.
Her development was delayed. She could sit without aid and crawl, but she could not independently walk at the age of 2 years and spoon herself. She was able to make eye contact, smile socially, understand simple instructions, and point out what she wanted. However, she could speak only in monosyllables. Overall, at the age of 2 years, the patient’s motor development was equivalent to approximately 10 months, although her cognitive and language abilities were relatively intact, at approximately 15 months.
Laboratory test results showed carbon dioxide retention on arterial gas analysis (68 mm Hg [normal range, 35 to 45]) and increased ammonia level (155 μg/dL [normal range, 12 to 66]). Except these, laboratory test showed normal complete blood count and normal chemistry test including creatine kinase level (36 IU/L [normal range, 32 to 135]). Chest computed tomography, laryngoscopy, cardiac echocardiography, and cardiac magnetic resonance imaging (MRI) revealed no abnormalities, except a small atrial septal defect. Genetic tests for myotonic dystrophy, spinal muscular atrophy, Prader-Willi syndrome, and Williams syndrome were performed, and all reports were negative. The patient’s beta-glucocerebrosidase level was assessed, and a metabolic workup was subsequently performed, including plasma amino acid, urine amino acid, serum lactic acid, and pyruvic acid. All test results were within the normal ranges, except those of serum lactic acid (3.22 mmol/L [normal range, 0.5 to 2.2]) and pyruvic acid (0.167 mmol/L [normal range, 0.034 to 0.102]). Electroencephalography and brain MRI were performed; however, no abnormalities were identified. She was subsequently referred to another hospital around her home for a follow-up.
At the age of 4 years, the patient was readmitted to our PICU due to respiratory failure. She showed delayed development, muscle weakness with no diurnal variation, and failure to thrive as well as mild ptosis with no diurnal variation and scoliosis with a Cobb angle of 51.0 degrees at T5–T9, which were not observed at the age of 2 years. We performed electromyography and nerve conduction velocity tests. Significant decremental response was observed in the 3-Hz repetitive nerve stimulation test. Subsequently, next-generation sequencing of neuromuscular disease was performed, and the patient was diagnosed with COLQ-related CMS (compound heterozygous mutation in the COLQ gene, p.Arg315Ter and p.Arg236Ter) (Table 2).
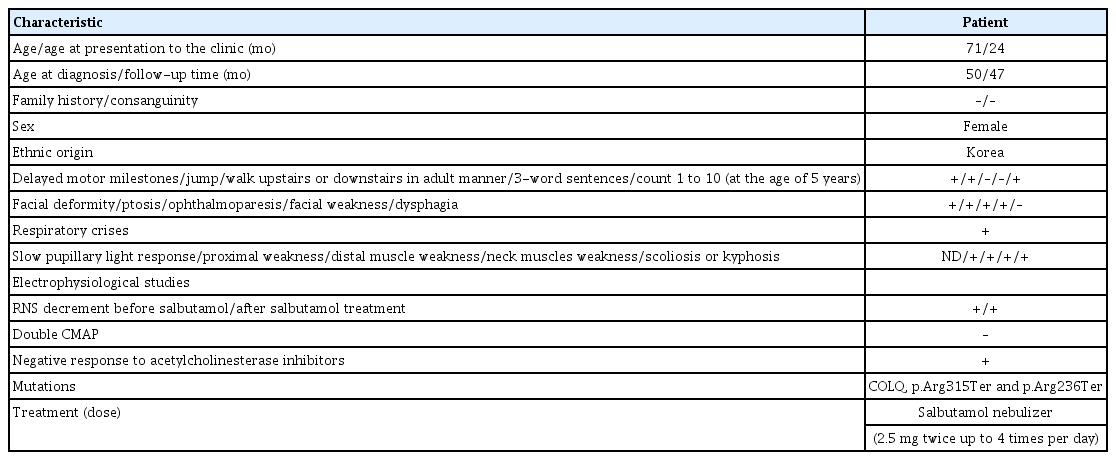
Genetic and demographic characteristics of patient in our case
After diagnosis, we began treatment with a salbutamol nebulizer (2.5 mg, twice per day) due to the absence of oral salbutamol and have maintained it for 2 years. Subsequently, her motor skills improved significantly. Her myasthenia gravis score improved from 7/39 (1 month after salbutamol treatment) to 14/39 (14 months after salbutamol treatment). In the 6-minute walking test performed, her walking distance was 130 m, and she was able to walk for 5 minutes (4 months after salbutamol treatment). We rechecked the same test at 14 months after salbutamol treatment; her walking distance was 135 m, and she could walk for 6 minutes. In particular, after beginning salbutamol treatment, she has not been admitted to our hospital and has not shown relapse or need for hospitalization in the PICU. Encouraged by these results, we increased the number of salbutamol treatments from two to four per day, and there have been no side effects so far.
To the best of our knowledge, this was the first reported case of the rare CMS in the Republic of Korea. When physicians encounter a patient with recurrent respiratory failure, they investigate the cause of the symptoms in the cardiovascular, pulmonary, and central nervous systems. However, as in our case, there may be other causes of recurrent respiratory failure. Because accurate diagnosis and appropriate evidence-based treatment can improve the quality of life in patients with CMS, we recommend that healthcare providers, specifically PICU physicians, consider recurrent respiratory failure as one of the clinical presentations of CMS and that they perform the relevant genetic evaluation. If COLQ-mutant CMS is confirmed, physicians should try to treat the patients using a salbutamol nebulizer.
This study was approved by the Yonsei University College of Medicine Institutional Review Board and the Research Ethics Committee of Severance Hospital (study approval number: 2019-3673-002). Written informed consent by the patients was waived due to a retrospective nature of our study.
Notes
Hoon-Chul Kang is an associate editor, Se Hee Kim is an editorial board member of the journal, but They was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contribution
Conceptualization: HGK and SHK. Data curation: HGK. Visualization: HGK and SHK. Writing-original draft: HGK. Writing-review & editing: HGK, JSL, KWK, HCK, and SHK.