Deep Phenotyping in 1p36 Deletion Syndrome
Article information

Abstract
Purpose
Although 1p36 deletion syndrome is the most common terminal deletion syndrome, unexplained phenotypic variability still occurs. We aimed to delineate the phenotype of this syndrome in detail and to characterize the phenotype-genotype correlation.
Methods
We retrospectively reviewed 15 patients diagnosed with 1p36 deletion syndrome confirmed by chromosomal microarray.
Results
All 15 patients revealed delayed attainment of motor milestones and speech. Seven patients (46.7%) never walked alone and only two (13.3%) could express a simple two-word sentence. They all showed subsequent intellectual disability. Two patients with large deletions of both distal and proximal critical regions of the 1p36 region shared severe intellectual disability with Rett syndrome-like behavioral features. Seizures, although frequent (73.3%), were well-controlled except in one patient with infantile spasms. Facial dysmorphism (92.9%) and ventricular mild dilatation with corpus callosum anomaly (46.7%) were common. Heart problems were identified in 14 patients, including structural abnormalities and/or functional problems associated with the gene encoding PR domain-containing protein 16. Two patients developed severe cardiac dysfunction requiring heart transplantation in their late teens. One patient with a 400 Kb deletion partly overlapping with the gene encoding calmodulin-binding transcription activator 1 did not have facial dysmorphism and presented with mild developmental delay and ataxic gait. One patient had a choledochal cyst, which was resected due to neonatal cholestasis.
Conclusion
Although the phenotype of 1p36 deletion syndrome is quite consistent with previous reports, additional manifestations such as certain behavioral features, ataxic gait, and severe cardiac dysfunction at an early age should be considered.
Introduction
The 1p36 deletion syndrome (OMIM 607872, also referred to as monosomy 1p36 syndrome) is the most common chromosome terminal deletion syndrome. This syndrome has some notable features. The estimated prevalence ranges from 1 in 5,000 to 1 in 10,000 [1-3]. Previous reports described the characteristic features of 1p36 deletion syndrome, which include developmental delay/intellectual disability (ID), epilepsy, craniofacial anomaly, and structural or functional heart problems [1,4,5]. However, some unexplainable phenotype variability still occurs. It is important to delineate clinical and genetic heterogeneity in terms of earlier recognition of the disorder, individualized surveillance testing for co-morbidities, and counseling on the prognosis. Recent advanced chromosomal microarray (CMA) enables physicians to detect interstitial deletions located in the far proximal site, identify complex rearrangements of the deletion site, and measure the extent of the deletion, which can precisely identify the involved genes compared to G-banded chromosomal analysis or telomere fluorescence in situ hybridization. We report our patients’ phenotypic spectrum of the 1p36 deletion syndrome with noticeable findings along with the exact breakpoint of chromosomes as detected by CMA. We also attempted to compare our results to previously reported findings in other literature.
Materials and Methods
Fifteen patients diagnosed with the 1p36 deletion syndrome by using CMA were retrospectively reviewed. The CMA test was conducted using Agilen Human Genome oligonucleotide comparative genomic hybridization microarray 244, 80, or 60 K (Agilent Technologies, Santa Clara, CA, USA) with 8.9, 13, or 41 Kb overall median probe spacing, respectively. All copy number variants (CNVs) were called and based on human assembly GRCh37 (hg19). Thirteen patient’s CMA data with exact chromosome deletion size and location were demonstrated with involved genes. Two patients’ clinical characteristics without detailed CMA data were also reviewed in attempt to review the phenotype variability.
Patients’ clinical characteristics and ancillary tests were retrospectively reviewed. Available cerebral magnetic resonance imaging (MRI), electroencephalography, echocardiography, abdominal ultrasonography, and laboratory results were all reviewed. Phenotypes with related genetic breakpoints and responsible genes described in previous literature were analyzed.
This study was approved by the Institutional Review Board of Seoul National University hospital (IRB No: H-2005-143-1125). Informed consent was waived by the board.
Results
1. Patients’ demographics
Of the 15 patients we recruited into the study, nine were female and six were male, giving a female to male ratio was 3:2. The median age at diagnosis was 4 years and ranged from 2 months to 15 years. The median follow up duration was 4.5 years and ranged from 0.8 to 9.9 years. All patients were aged more than 4 years and four patients were over 18 years old at the last follow-up. Eight patients’ parental tests were available and all were reported as de novo deletions in the 1p36 region. All 15 patients’ demographic data and overall clinical features are provided in Table 1.
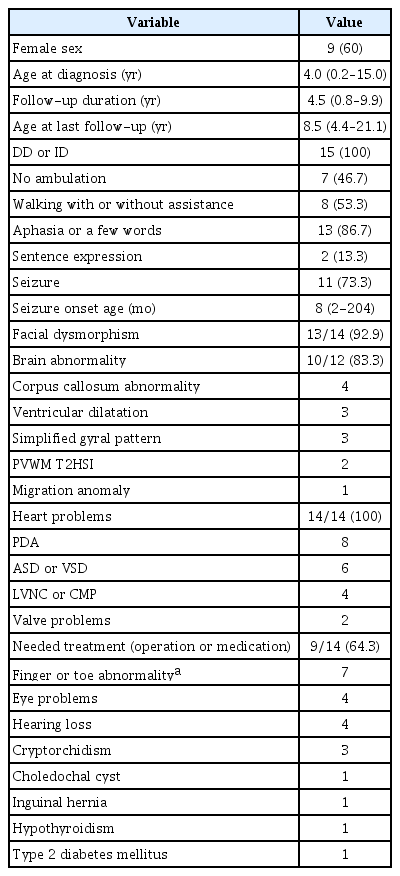
Overview of the clinical features of the 15 patients with 1p36 deletion (n=15)
2. Neurological problems
All 15 patients were reported to have varying degrees of developmental delay or ID. Delayed attainment of motor milestones and speech was apparent in all 15 patients. Although all patients could sit with or without support eventually, seven patients (46.7%) were not able to get walking ability even after their age of 4 years. Those patients also showed limited language acquisition. At the most, a few word expression was possible. Eight patients (53.3%) could walk with or without assistance; however, only two patients were able to express a simple sentence consisting of two basic words. One patient (Patient 6) with the smallest chromosome deletion (400 Kb, chr1:6499296-6900414) who presented with slow catch-up development could walk alone at his age of 31 months and revealed the mildest ID among 15 participants. Other 14 patients showed severe ID without developmental regression (Table 2).

Neurologic characteristics of the 15 patients with 1p36 deletion syndrome
A noticeable feature was found in two relatively older children aged 10 and 13 years that visited (Patient 2 and 3, respectively). They presented with Rett syndrome-like behavioral features including severe ID with hand automatism or bruxism showed large deletions that overlapped both distal [6] and proximal [7] critical regions. Other behavioral features such as aggressive behavior (Patient 5) and attention deficit hyperactivity disorder (Patient 6) were also described. The available CMA data of thirteen patients are depicted in Fig. 1.
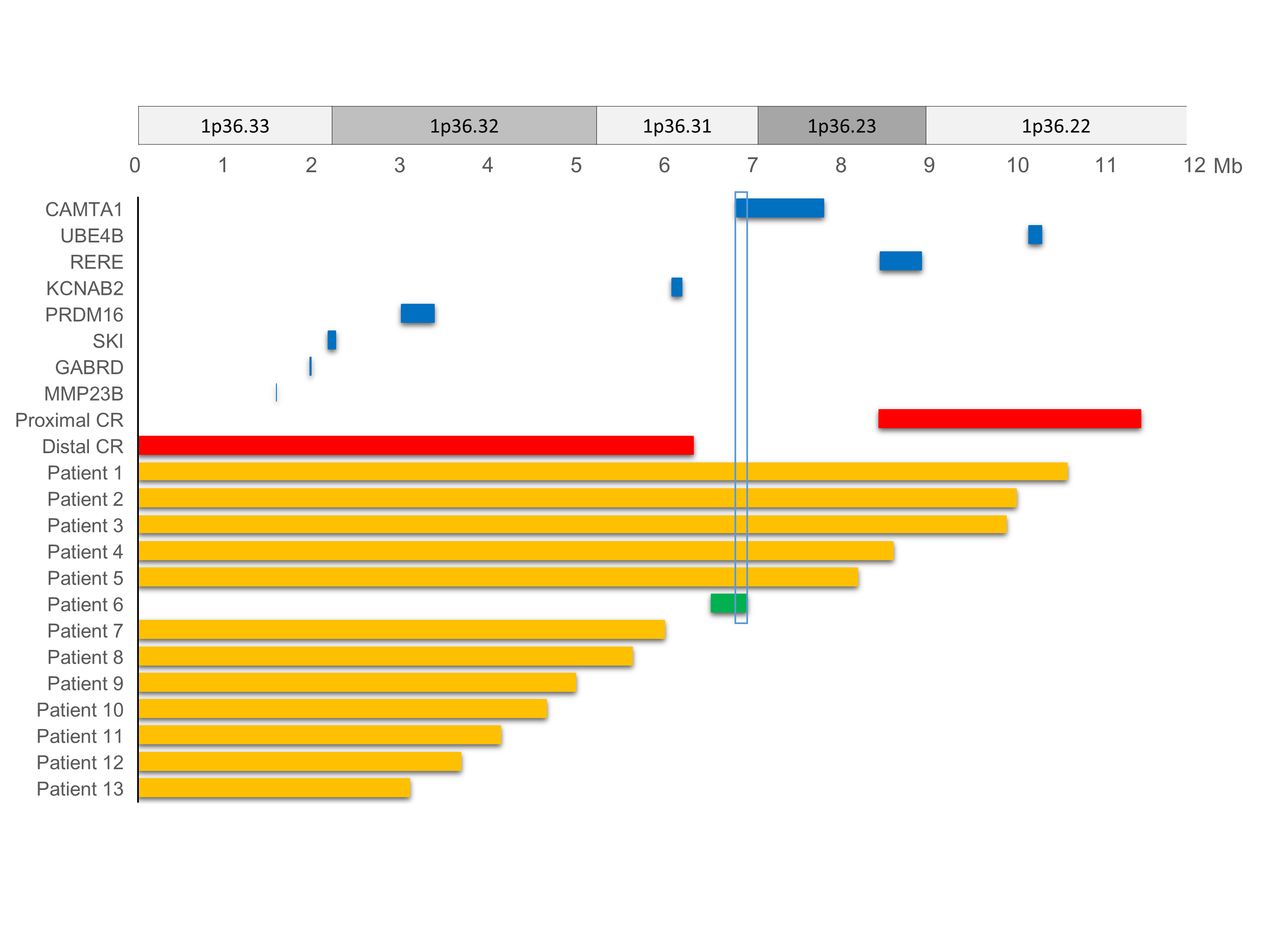
Chromosomal terminal deletion of the 1p36 region in 13 patients with candidate gene locations. Thirteen patients’ chromosome breakpoint and location data are depicted based on available chromosomal microarray test results. Each solid bar represents the deletion point and size. Interstitial deletion is illustrated with a solid green bar. Previously reported proximal/distal critical regions (CRs) and known candidate genes are shown in solid red and blue color bars, respectively. Calmodulin binding transcription activator 1 (CAMTA1; chr1: 6,845,384-7,829,766) was partially deleted (chr1: 6845384-6900414) in patient 6. Other previously reported candidate genes are also depicted with their size and location. UBE4B, ubiquitination factor E4B; RERE, arginine-glutamic acid dipeptide repeats; KCNAB2, potassium voltage-gated channel subfamily A regulatory beta subunit 2; PRDM16, PR domain-containing protein 16; SKI, SKI proto-oncogene; GABRD, gamma-aminobutyric acid receptor delta; MMP23B, matrix metalloproteinase 23B.
Seizures occurred in 11 patients (11/15, 73.3%). Every first-time seizure occurred before the age of five except in one patient (Patient 2, seizure onset at 17 years old). In seven patients (63.6%), seizure was noticed before the age of one and the earliest onset age was 2 months. Seizure semiology was variable, including focal clonic, generalized tonic or clonic, hypomotor, or spasm. Only one patient (Patient 1) had infantile spasms and was detected hypsarrhythmia in electroencephalography. Combination of vigabatrin, zonisamide, and prednisolone were required in this patient whose deletion size was the largest one (10.5 Mb) of all the study participants. Except in this patient, seizures were well-controlled with zero (n=2) to two (n=2), mostly one (n=6), antiepileptic drug (AED). Three patients were able to discontinue AED after 3 years from their initial treatment. There was no correlation between seizure onset age and seizure severity.
3. Facial dysmorphism
Facial dysmorphism was noticed in 13 patients with flat and pale faces, deep-set eyes, small-slit eyes, hypertelorism, flat noses and nasal bridges, low set ears with small or large cupped ears, thickened ear helices, small mouths, or pointed chins. Although some previously delineated dysmorphic facial features were noticed, we could not identify any shared facial anomaly between the patients. There was no relationship between the deletion sizes and facial dysmorphism. However, one patient (Patient 6) with the smallest size of deletion (400 Kb) did not have any apparent craniofacial anomaly.
4. Brain structural abnormality
Brain structure was able to assess in 12 patients with MRI. Among them 10 patients (83.3%) had some abnormalities including four patients with corpus callosum abnormalities (hypoplasia or dysgenesis), three patients with ventricular dilatation, three patients with simplified gyrus patterns, two patient with periventricular white matter T2 high signal intensity, and one patient with migration anomaly. Two patients including one patient (Patient 6) with the smallest size of interstitial deletion (400 Kb) did not have any brain structural abnormality.
5. Heart problems
Except for one patient who did not undergo echocardiography, all patients had a certain degree of heart anomalies or functional problems. Patent ductus arteriosus was the most common problem (57.1%) followed by atrial septal defect (ASD) or patent foramen ovale (35.7%), ventricular septal defect (VSD; 28.6%), left ventricular noncompaction (LVNC), or dilated cardiomyopathy (DCMP; 28.6%), and valve anomaly (14.3%; severe mitral valve stenosis, bicuspid aortic valve). Nine patients required either corrective operations for cardiac anomalies or pharmacological treatments for cardiac dysfunction (9/14, 64.3%). Of the three patients (Patient 7, 11, and 13) with DCMP, two patients (Patient 11 and 13) had end-stage cardiac dysfunction impending heart transplantation at their ages of 19.6 and 19.0 years, respectively. All four patients with either LVNC or DCMP revealed that their deleted chromosome sites were involved with the gene encoding PR domain-containing protein 16 (PRDM16), previously well known as one of the major genes responsible for cardiomyopathy. Patient 6, with the smallest interstitial deletion (400 Kb), had ASD and VSD. However, those defects did not require any treatment, and closed spontaneously.
6. Other phenotypes
Almost half of the patients (7/15, 46.7%) had either finger or toe anomaly. The phenotypic descriptions were either overriding, curly, or contracture deformity of fingers or toes. Amblyopia or esotropia was also found in four patients. Sensory neural hearing loss was another problem in four patients. Cryptorchidism (n=3), inguinal hernia (n=1), hypothyroidism (n=1), and type 2 diabetes mellitus (n=1) were reported as well. A choledochal cyst with neonatal cholestasis was observed in Patient 12. None of them was reported to have neuroblastoma. The patients’ non-neurological clinical phenotypes were provided in Table 3.

Detailed non-neurologic phenotypes of the 15 patients with 1p36 deletion syndrome
Discussion
In patients with the 1p36 deletion syndrome, a certain phenotype with a responsible deleted region or a causative gene was still under investigation along with cumulating CMA data [5,8]. Although deletion size is not linearly correlated with the clinical severity, critical regions and involved genes in 1p36 region were reported in previous studies [7-10].
Moderate to severe ID is present universally in the patients with 1p36 deletion syndrome [1]. Shimada et al. [8] described the genomic regions responsible for ID and associated possible modifier genes, potassium voltage-gated channel subfamily A regulatory beta subunit 2 (KCNAB2; chr1:6,052,358–6,161,253; OMIM# 601142). They reported that patients with terminal deletions larger than 6.2 Mb from telomere showed no ambulation and poor prognosis of neurodevelopmental status. We noticed the similar findings in our five patients (Patient 1 to 5) with larger deletions including KCNAB2 gene. They were having severe neurocognitive delay prominent with speech delay and not able to gain walking ability at last follow-up in their age of 7, 20, 21, 8, and 5 years, respectively. Patient 14 (4 years old) and 15 (5 years old) were not able to walk at their last follow-up; however, no detailed information of deletion size was available. We could assume that they might have larger than 6.2 Mb deletions.
Behavioral problems were not easy to be specified due to severe ID. However, an unknown cause of female ID with Rett syndrome like behavioral features could be another behavioral phenotype of the 1p36 deletion syndrome. Patients 2 and 3 presented with severe ID with hand automatism, bruxism, and abnormal breathing pattern. It was clinically indicated Rett syndrome. In contrast to Rett syndrome, they showed early onset developmental delay without regression and mild anomalies in face, fingers, or brain. Patients 2 and 3 were tested negative for the MECP2 deletion/duplication and had large deletions of 9.9 and 9.8 Mb, respectively in the 1p36 region. This finding is another possible behavioral expression of the 1p36 deletion syndrome along with the Prader-Willi syndrome-like behavioral expression [11,12]. Further investigation of genes responsible for behavioral expressions should be performed in the future study.
Seizures were a common manifestation reported from 50% to 79% [1,4,8]. Infantile spasms and refractory epilepsy (20.9%) were associated with poor clinical outcomes in a previous study [13]. Our study also showed a high rate of occurrence of seizures (11/15, 73.3%). However, except for one patient presenting with infantile spasms having the largest deletion size, seizures were well-controlled, or even resolved spontaneously. Four patients were required AED at last follow-up and their deletion size were over 5.9 Mb. Although it demands a larger cohort study to establish statistical significance, the severity of seizure seems to be related to the size of the deletion rather than to the onset age of the seizure.
We noted that each patient had some shared facial appearances with reported features. However, not all patients were able to be suspicious of the 1p36 deletion syndrome based on facial features only. There was no specific pattern of congenital brain structural abnormality either. One patient (Patient 6) with a small interstitial deletion of 400 Kb (chr1:6499296-6900414) showed a normal facial appearance and brain structure.
Cardiac assessment is crucial at initial evaluation and also during long term follow-up period in 1p36 deletion syndrome. Structural anomaly with or without left ventricular dysfunction is a major concern in 1p36 deletion syndrome patients in view of their long term prognosis. Previous literature reported heart problems as prevalent manifestations with up to 75% of structural anomaly and 23% to 31% of cardiomyopathy [4,5,14]. Our report showed the possible severity of cardiomyopathy as an end-stage severe heart dysfunction in two patients. These two patients were impending heart transplantation as they were approaching adulthood. Cardiac dysfunction in two other patients was not severe; however, the older patient required medications at the age of 10 years during the last follow up. Due to well-controlled seizures in early childhood and absence of symptoms with slowly progressing heart dysfunction, follow up might be irregular, and as such early prophylactic medical treatment may be missed. Encouraging regular follow-up with explanations of possible heart involvement in 1p36 deletion syndrome in late teenage should be included in patients’ and parents’ education, even in a patient without any symptom or sign.
One patient (Patient 6) with a small interstitial deletion of 400 Kb (chr1:6499296-6900414) was presented with relatively mild phenotype, including mild global developmental delay, ataxic gait, unilateral crytorchidism, amblyopia, and spontaneously-closed ASD and VSD. Brain MRI was normal and he had no history of seizure. The deleted position revealed 11 involved genes including three high pLI (loss of function intolerance) genes; PHD finger protein 13 (PHF13), DnaJ heat shock protein family member C11 (DNAJC11), calmodulin-binding transcription activator 1 (CAMTA1). Among them, CAMTA1 was previously reported as the cause of non-progressive congenital ataxia with or without ID [15,16], which was consistent with the phenotype of this patient having mild global developmental delay and unexplainable ataxic gait. This deleted region was not regarded as a critical region in the previous literature. This patient showed amblyopia and cryptorchidism as well. However, there was no shared deleted region among patients who had amblyopia and cryptorchidism. This finding might be another explanation for the shared phenotype with different involved genes [17].
The 1p36 deletion syndrome showed a tendency of contiguous gene deletion syndrome presenting a more severe phenotype in case of larger deletion size. However, there was often discordance between deletion size and clinical features. Even cases with small deletion sizes had most of the clinical features of 1p36 deletion syndrome [4]. It is a reasonable assumption that the distal part of the 1p36 region is responsible for distinct features of the 1p36 deletion syndrome. Beyond this very critical terminal region, continuous efforts to delineate phenotype variability with exact genotyping is crucial in a variable range of deletion sizes, and an inconsistent breakpoint of the chromosome 1p36 region.
Notes
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: YS, YJG, SYK, HK, HH, JC, BCL, KJK, and JHC. Data curation: YS, YJG, SYK, HK, HH, JC, BCL, KJK, and JHC. Formal analysis: YS, SYK, and JHC. Methodology: YS, SYK, and JHC. Project administration: YS. Visualization: YS. Writing-original draft: YS. Writing-review & editing: YS and JHC.