Diagnosis of Tuberous Sclerosis Complex and Epilepsy Outcomes in Children with Fetal Cardiac Rhabdomyoma: A Long Term Follow-up Study
Article information

Abstract
Purpose
Prenatal diagnosis of cardiac rhabdomyoma is suggestive of the presence of tuberous sclerosis complex (TSC), which is commonly associated with epilepsy. This study investigated the diagnostic rate of TSC, the incidence and treatment outcomes of epilepsy, and long-term neurodevelopmental outcomes in children with fetal cardiac rhabdomyoma.
Methods
We retrospectively reviewed the medical records of 50 patients with fetal cardiac rhabdomyoma between 1995 and 2017 at the Asan Medical Center Children’s Hospital. Twelve patients were excluded because of incomplete data or a follow-up of less than 1 year. Patients’ medical records were assessed for the occurrence of epilepsy during follow-up, including treatment and outcomes, electroencephalography and magnetic resonance imaging, genetic analysis findings, and neurodevelopmental status.
Results
Thirty-eight patients (23 males) were diagnosed with TSC. Twenty-eight (73.7%) developed epilepsy. Infantile spasms were observed in 22 patients (78.6%), and 21 (95.5%) received vigabatrin as the first treatment, achieving subsequent freedom from seizures. At the final evaluation, 17 patients (60%) were seizure-free for over 12 months. Seizure remission was not associated with seizure onset age, the presence of cortical tubers, or TSC1/2 mutations. Patients with epilepsy had a higher prevalence of poor neurodevelopmental outcomes than did those without epilepsy (P=0.008).
Conclusion
A high TSC diagnostic rate and a high incidence of early-onset epilepsy were observed in patients with prenatal cardiac rhabdomyoma, and children with TSC and epilepsy showed poor neurodevelopmental outcomes. Therefore, monitoring with electroencephalography follow-up of infants with fetal cardiac rhabdomyoma may be helpful for the early detection and treatment of epilepsy.
Introduction
Tuberous sclerosis complex (TSC) is a multi-systemic, autosomal dominant disorder affecting various organs, including the brain, heart, kidneys, liver, eyes, lungs and skin. It is estimated that TSC occurs in about 1 out of 6,000 live births [1].
In fetuses with TSC, cardiac tumors and cortical tubers can be detected in the prenatal period [2-4]. As multimodal imaging has improved, TSC can be diagnosed prenatally if there are any of the following: cardiac rhabdomyomas, cortical tubers, subependymal nodules, and renal angiomyolipomas [5]. However, the presence of fetal cardiac rhabdomyoma alone is not diagnostic of TSC and the diagnostic rate of TSC in patients with fetal cardiac rhabdomyoma is still unknown [6,7].
Epilepsy is the most common neurologic manifestation in patients with TSC with an incidence of 80% to 90% during the first year of life, with infantile spasms being a very common epilepsy phenotype [4,8]. Early onset and intractable seizures pose a considerable risk of neurodevelopmental and cognitive problems [9-11], and early and immediate seizure control is important in preventing the development of epileptic encephalopathy and intellectual disability [12]. Therefore earlier diagnosis of epilepsy can enable earlier treatment and improve outcomes in children with TSC [10].
This is a retrospective study with long-term follow-up on the natural course of epilepsy and developmental outcomes of patients prenatally diagnosed with cardiac rhabdomyoma. We aimed to describe diagnostic rate of TSC, incidence and treatment outcome of epilepsy, related clinical factors of seizure remission, and long-term neurodevelopmental outcomes in patients prenatally diagnosed with cardiac rhabdomyoma.
Materials and Methods
1. Patients
Between 1995 and 2017 in Asan Medical Center Children’s Hospital, 50 patients were prenatally diagnosed with cardiac rhabdomyoma. Of those, 12 patients were excluded because of incomplete medical record data or short-term follow-up less than 1 year and 38 patients were included in this study.
2. Data collection
Of 38 patients, all clinical data were included through March 2017. From our electronic clinical database, we extracted data including demographics (gestational age at birth, birth weight, sex, family history), other TSC manifestations, testing for TSC1 and TSC2 mutation, diagnosis of epilepsy, date of seizure onset, seizure types, presence of hypsarrhythmia, epilepsy treatment (including type and total number of anti-epileptic drugs [AEDs] prescribed during follow-up periods, and non-pharmacologic treatments such as epilepsy surgery, vagus nerve stimulation, and ketogenic diet), and developmental outcomes. The TSC diagnosis followed the recent recommendations from the Tuberous Sclerosis Consensus Conference in 2012 [13]. All patients regularly underwent a complete medical examination, with electroencephalogram (EEG), magnetic resonance imaging (MRI), abdominal sonography, and cardiologic/ophthalmic tests. Data on cortical tubers, cardiac complications, and associated anomalies were collected from the final follow-up evaluation. EEG findings at initial seizure onset were classified according to the presence or absence of hypsarrythmia. Seizure freedom was defined as no seizures occurring for at least 1 year. The definition of well-controlled epilepsy is seizure free at least 1 year at the time of the last evaluation and the definition of uncontrolled epilepsy is sustained seizures within 1 year at final evaluation.
Neurocognitive assessment results were available for all 35 of 38 patients. Abilities were assessed using the Korean Infant and Child Development Test (KICDT; n=24, 68%) and Bayley Scales of Infant Development (BSID-II; n=4, 11%). Children who were older than 5 years of age were tested with the Korean Educational Development Institute-Wechsler Intelligence Scale for Children (KEDI-WISC; n=7, 20%). Those with an intelligence quotient <70 were considered to have intellectual disability and those with a development quotient <80 were considered to be delayed in their development. This study was approved by the Institutional Review Board at the Asan Medical Center (IRB No. 2019-1076). Written informed consent by the patients was waived due to a retrospective nature of our study.
3. Data analysis
The independent samples T-test and Mann-Whitney test were used to compare the sizes of the two independent groups of outcome variables. Chi-square tests were used for categorical data regarding the assessment of associated clinical risk factors by using SPSS version 21.0 (IBM, Armonk, NY, USA).
Results
1. Patients characteristics
All 38 patients (23 males and 15 females) who had cardiac rhabdomyoma identified by prenatal sonography were diagnosed with TSC during the follow-up period because they met two major clinical features: cardiac rhabdomyoma and skin manifestations of TSC. Some of them were also diagnosed as TSC with genetic analysis: a mutation of the TSC1 gene was found in three of 38 patients (7.9%) and a mutation in TSC2 in 15 of 38 (39.5%). The median follow-up duration was 4.5 years (range, 1.3 to 26). Five patients (13.2%) were born prematurely (GA <37 weeks). Most cortical tubers identified on MRI were diffuse, multifocal type (89.5%). Some patients had cardiac complication: six patients (15.8%) had arrhythmia such as premature ventricular contractions, paroxysmal supraventricular tachycardia, Wolff-Parkinson-White syndrome and two patients (5.3%) had structural anomalies such as left ventricular outflow tract obstruction (LVOTO), tetralogy of fallot (TOF). Patient with LVOTO had partial tumor resection operation and patient with TOF had TOF correction operation because of their structural anomalies. Spontaneous tumor regression occurred in 10 patients (35.7%). Baseline demographics are shown in Table 1.
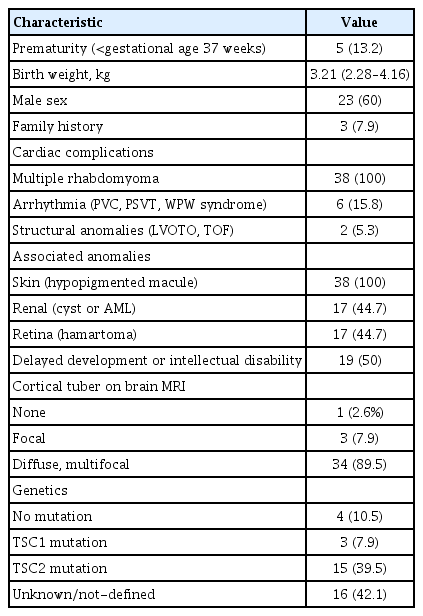
Patient characteristics (n=38)
2. Epilepsy characteristics and related clinical risk factors
With a median follow-up of 4.5 years (range, 1.3 to 26), 28 patients (73.7%) developed epilepsy and mean age at seizure onset was 9 months (range, 0.13 to 32). At the time of the last evaluation, 17 patients (60.7%) were seizure-free for longer than 12 months. Infantile spasms were observed in 22 patients (78.6%), 21 of whom (95.5%) received vigabatrin as the first treatment and achieved subsequent seizure freedom. Epilepsy characteristics are shown in Table 2.
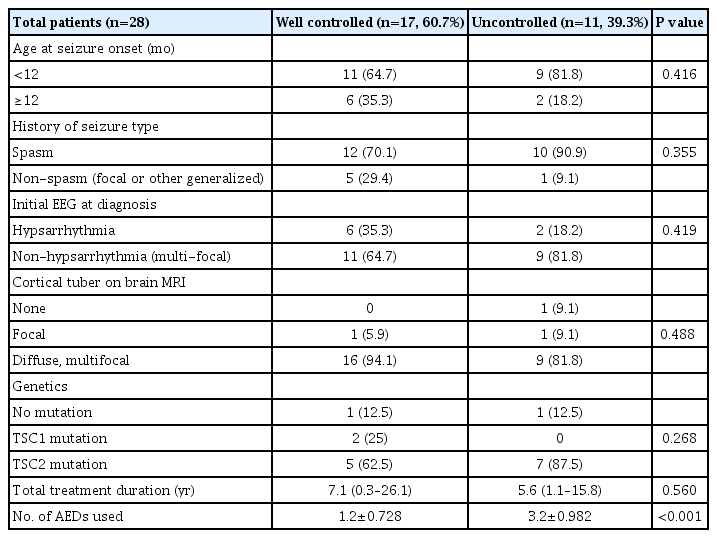
Comparison of clinical characteristics between well-controlled and uncontrolled epileptic patients with tuberous sclerosis complex
Table 2 also shows the comparison of clinical characteristics between patients with well-controlled epilepsy (seizure free at last evaluation) and patients with uncontrolled epilepsy (sustained seizures at final evaluation). Among 28 patients who developed epilepsy, there were 17 (60.7%) with well-controlled epilepsy and 11 (39.3%) with uncontrolled epilepsy at the final evaluation. Apart from the number of AEDs used, there was no difference in clinical characteristics between well-controlled and uncontrolled patients (1.2±0.728 for well-controlled patients, 3.2±0.982 for uncontrolled patients, P<0.001). Seizure remission was not associated with age at seizure onset, the presence of cortical tubers, or TSC1/2 mutations. One patient received vagus nerve stimulation (n=1) as a non-pharmacologic treatment.
A total of 14 different AEDs were used and some patients received multiple AEDs. Fig. 1 shows AED types used between patients with well-controlled and uncontrolled epilepsy. The most common AEDs among patients with well-controlled epilepsy were vigabatrin (n=13), and zonisamide (n=5), while patients with uncontrolled seizure received clobazam (n=8), oxcarbazepine (n=6), topiramate (n=6), and rufinamide (n=2).
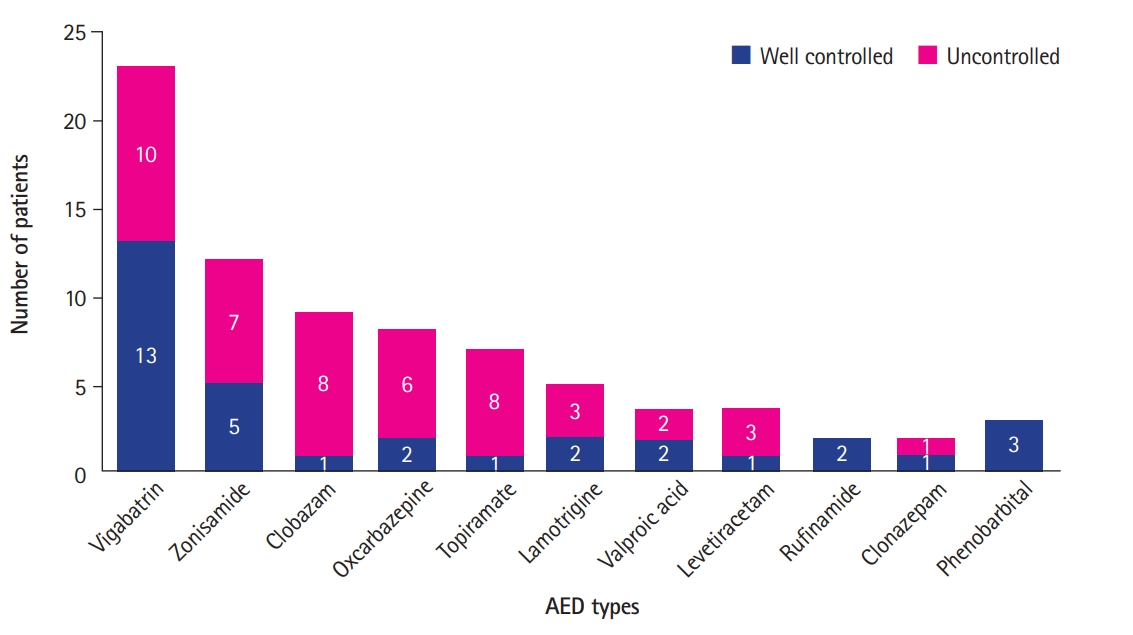
Anti-epileptic drugs (AEDs) used in patients with tuberous sclerosis complex and epilepsy whether seizures were well-controlled or uncontrolled. A total of 14 different AEDs were used and some patients received multiple AEDs. The most common AEDs among patients with well-controlled epilepsy were vigabatrin (n=13), and zonisamide (n=5), while patients with uncontrolled seizure received clobazam (n=8), oxcarbazepine (n=6), topiramate (n=6), and rufinamide (n=2).
3. Neurodevelopment outcome
With a median follow-up of 4.5 years (range, 1.3 to 26), 19 of 38 (50%) patients were identified as having delayed development or an intellectual disability (Table 1). Eighteen of 28 patients (64.3%) with epilepsy were found to have delayed development or intellectual disability. Among the 10 patients without epilepsy, a smaller proportion (10%) had delayed development or intellectual disability (Fig. 2). Epileptic patients were more likely to have poor neurodevelopmental outcome (P=0.008). Among epileptic patients, children with delayed development had longer periods of sustained seizure (0.44±0.578 years for children with normal development, 4.12±4.468 years for children with delayed development, P=0.018).
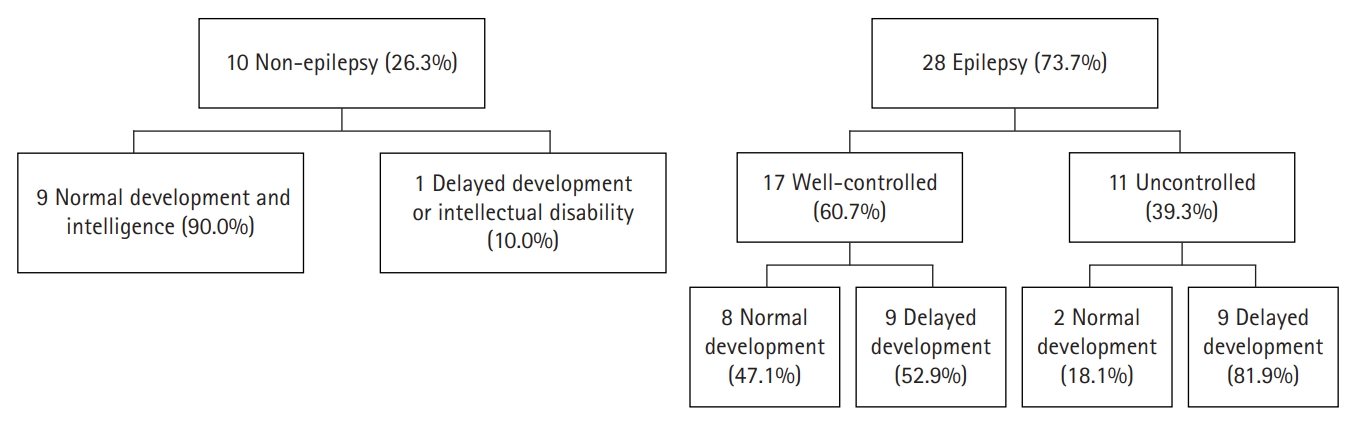
Neurodevelopmental outcomes in non-epilepsy group and epilepsy group. Nineteen of 38 (50%) patients were identified as having delayed development or an intellectual disability. Eighteen of 28 patients (64.3%) with epilepsy were found to have delayed development or intellectual disability. Among the 10 patients without epilepsy, one patients (10%) had delayed development or intellectual disability.
Discussion
TSC is an autosomal dominant congenital syndrome and appears in various phenotypes caused by mutations in one of the tumor suppressor genes, TSC1 or TSC2 [14]. TSC is known to have benign tumors in various tissues and neurological manifestations such as epilepsy, mental retardation, and behavioral problems [8]. In recent decades, with developing imaging techniques, TSC can be diagnosed in the prenatal period if there are any of the following: cardiac rhabdomyomas, cortical tubers, subependymal nodules, or renal angiomyolipomas [5]. Among those, multiple cardiac rhabdomyomas are the most common prenatal sign of TSC [2]. In this study, we selected patients diagnosed with fetal cardiac rhabdomyoma in a single tertiary hospital and analyzed their follow-up data. All 38 patients who had prenatal cardiac rhabdomyoma were eventually diagnosed with TSC during the follow-up period. Given the high rate of diagnosis of TSC following presentation with prenatal cardiac rhabdomyoma, it seems that more emphasis on the surveillance for other TSC features in these patients may be warranted [7].
Epilepsy is a common neurological symptom and this study also showed that TSC patients are at high risk for developing epilepsy [4,8]. In our study, 28 of 38 patients (73.7%) developed epilepsy with a mean age at seizure onset of 9 months (range, 0.13 to 32). This is similar figure as those given by Jozwiak et al. [15] and Wu et al. [16], and tuberous sclerosis registry to increase disease awareness (TOSCA) study on large population also reported that epilepsy developed in 86.3% of patients and 79.3% were diagnosed with epilepsy before 2 years [17]. In addition, a high incidence of infantile spasms (78.6%) was observed in this study, which is different from the result of the TOSCA study: 38.9% presented with infantile spasms and 67.5% with focal seizures [17]. Twenty-one of patients with infantile spasms (95.5%) received vigabatrin as the first treatment and achieved subsequent seizure freedom. At the TSC Consensus Meeting for Subependymal Giant Cell Astrocytoma and Epilepsy Management, the latest recommendations for treatment of epilepsy in TSC is vigabatrin as a first line monotherapy for both infantile spasms and focal seizures [18]. Vigabatrin is a highly selective inhibitor of gamma-aminobutyric acid (GABA) transaminase and epileptogenesis in TSC is associated with reduced activity of GABA inhibition. For this reason, vigabatrin is thought to be particularly effective against epilepsy caused by TSC [19-21]. Although use of vigabatrin was not associated with seizure freedom in this cohort, the use of vigabatrin should be considered in young children with TSC. Many previous studies showed early control of seizures might improve cognitive and behavioral outcomes [10,11,22]. Most of children in the cohort used vigabatrin as the first AED for their seizure control and achieved good outcomes. Visual field defects have been reported as a side effect of vigabatrin [23-25], but this has been reported to be less common in young children than in adults and are relatively safe for use in children [21].
In this study, seizure remission was not associated with age at seizure onset, the presence of cortical tubers, or TSC1/2 mutations, probably due to small number of patients. Previous study showed that patients with later onset seizures had longer periods of seizure freedom [12,22]. Samir et al. [14] observed that early seizure onset (<6 months) is related with poor seizure outcomes. Jansen et al. [26] reported that patients with TSC2 mutations have more intractable seizures than those with TSC1 mutations. An increased number of tubers has also been associated with poor seizure control [11,27]. Other studies, however, have suggested no significant association between higher tuber count and poor seizure control [28,29].
The rate of intellectual disability in patients with TSC is 40% to 70%, while severe intellectual disability has been reported as high as 30% to 45% of these patients [30-32]. A previous large series of 160 patients from Mayo Clinic also showed a strong association between intellectual disability and epilepsy [33]. In our cohort of patients who were prenatally diagnosed with cardiac rhabdomyoma, patients with epilepsy tend to have poor neurodevelopmental outcomes; this finding is consistent with earlier studies suggesting that epilepsy is an important risk factor for developmental delay [30-32,34,35]. Previous studies have shown that EEG abnormalities precede epilepsy onset in TSC patients [1] and improved developmental outcomes were observed when the anti-epileptic treatment was given to infants before the onset of clinical seizures [15]. Wu at al. [16] and Whitney et al. [36] also showed prospectively evaluated usefulness of EEG surveillance for prediction of epilepsy in TSC. This finding suggests that close EEG follow-up of patients with fetal cardiac rhabdomyoma can be used lead to better outcomes in these patients.
This study analyzed long-term follow-up data of a relatively large population of children with fetal cardiac rhabdomyoma, a rare disease. Due to the retrospective nature of this study, evaluation schedules, clinical follow-up, and developmental screening tests were variable between patients. Further, large proportion of the patients were lost to follow-up.
In conclusion, all patients in this study with prenatal cardiac rhabdomyoma were eventually diagnosed with TSC, either clinically or genetically during the follow-up period. Although it is controversial as to whether all patients who have prenatal cardiac rhabdomyoma will also be diagnosed with TSC, it is clear that these patients are high risk group for TSC and should be monitored for other clinical manifestations of TSC. In this study, epilepsy appears in 78.7% of patients with TSC, most commonly before first year of age. About 80% of children with epilepsy in TSC patients present with infantile spasms, most of them were treated with vigabatrin and had good responses like previous studies. In this cohort, development of epilepsy was also associated with poor neurodevelopmental outcomes. Therefore, all infants prenatally diagnosed with cardiac rhabdomyoma have high potential of being diagnosed with TSC and should be closely monitored with EEG follow-up for the early diagnosis and treatment of epilepsy.
Notes
No potential conflict of interest relevant to this article was reported.
Author contributions
Conceptualization: SJY, MSY, and TSK. Data curation: DK and HJK. Formal analysis: DK and HJK. Funding acquisition: MSY and TSK. Methodology: SJY, MSY, and TSK. Project administration: SJY, MSY, and TSK. Visualization: DK. Writing-original draft: DK. Writing-review & editing: SJY, MSY, and TSK.
Acknowledgements
This study was supported by a grant (2019IT0002) from the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Korea.
The authors would like to thank Scientific Publications Team of Asan Medical Center for providing language help.