De Novo SCN8A Pathogenic Variant (c.5630A>G; p.Asn1877Ser) Presenting with a Relatively Mild Phenotype
Article information

Voltage-gated sodium channels are composed of one pore-forming α subunit and one or two auxiliary β subunits. So far, nine voltage-gated sodium channel α-subunits (Nav1.1–Nav1.9) have been identified [1]. Among them, Nav1.6 is encoded by the SCN8A gene, and widely expressed in the projection neurons of the cerebral cortex, hippocampus, and cerebellum [2]. In general, pathogenic variants (PVs) in SCN8A associated with early onset epileptic encephalopathy are de novo missense variants, and electrophysiological studies of such PVs typically reveal ‘gain of function’ leading to enhanced sodium current [3]. Since the first report of SCN8A‐related epilepsy in 2012, more than 100 cases have been reported. SCN8A PVs mostly causes early onset epileptic encephalopathy type 13, which presents with infantile onset epilepsy accompanied with developmental delay and cognitive impairment [1]. However, recent reports have demonstrated that missense PVs in SCN8A present with mild phenotypes [1,2,4].
A 6-month-old male infant was admitted to the emergency department at Ulsan University Hospital due to seizure. Until the day before admission, the infant was healthy with no history of medication, head trauma, or recent immunization. Upon arrival at the emergency department, the patient’s seizure had already ceased. His body temperature was 37.1℃; heart rate was 158 beats/min; and respiratory rate was 26 breaths/min. His pupils were isocoric, and the light reflex of both eyes was prompt. One hour after arrival at the emergency department, another generalized tonic-clonic seizure developed, and it subsided soon after intramuscular administration of lorazepam. The patient was born at 40 weeks’ gestation via a cesarean section without complications. His growth and development was age appropriate. The patient’s parents had no history of neurological diseases, including epilepsy. He had no siblings. Laboratory results revealed that sodium was 136 mmol/L, total calcium 11.1 mg/dL, and glucose 99 mg/dL. Electroencephalography revealed normal finding. Magnetic resonance imaging of the brain showed no abnormalities except a small developmental venous anomaly in right frontal lobe. On day 1, the patient showed a third generalized tonic-clinic seizure in the pediatric ward, and it was managed with intravenous phenobarbital. He had no additional seizure afterward, and was discharged on day 4. After discharge, the patient maintained oral phenobarbital administration (5 mg/kg/day) without further seizure for 2 months. However, at the time of a phenobarbital taper, the patient developed two additional generalized seizures lasting about 1 minute within 3 hours, and was readmitted to the emergency department. During the second hospitalization, he had 10 additional seizures, all of which were generalized tonic-clinic seizures lasting between 15 seconds and 15 minutes. Intravenous valproate and levetiracetam, and oral clobazam failed to control the seizures. The patient’s seizures were eventually controlled with commencement of oxcarbazepine administration on day 15. He was discharged on day 21 with discharge medications of oxcarbazepine, valproate, and clobazam. For molecular diagnosis, we performed targeted gene sequencing of 118 epilepsy-related gene panel at GC genome (Yongin, Korea), which revealed two missense variants in SCN8A (NM_014191.3:c.5630A>G; p.Asn1877Ser) and CHRNB2 (NM_000748.2:c.691C>T; p.Arg231Cys). The SCN8A variant has not been observed in the population databases, but has previously been reported in patients with epilepsy (PMID: 27210545, 27875746, 29100083, 27864847, 28923014, 31487502, 31164858, 30851583, 30776697). On the other hand, the population frequency of the CHRNB2 variant was estimated to be 0.0028% in the population database (gnomAD; https://gnomad.broadinstitute.org/), but has not been reported in patients with epilepsy. By Sanger sequencing of the SCN8A and CHRNB2 variants in the patient and his parents, the SCN8A variant was not observed in both parents while the CHRNB2 variant was observed in his father confirming de novo occurrence of the SCN8A variant (Fig. 1). Although the CHRNB2 gene is known to be associated with autosomal dominant nocturnal frontal lobe epilepsy, the patient’s father never had a seizure in his life and had no neuropsychological problems. Hence the CHRNB2 variant was regarded as likely benign. After the second discharge, clobazam was discontinued, and oxcarbazepine (25 mg/kg/day) and valproate (20 mg/kg/day) was maintained. So far, the patient has remained seizure free for more than 6 months. He has reached normal developmental milestones, with saying “Mama” and “Papa” at 13 months of age and walking unaided at 14 months of age.
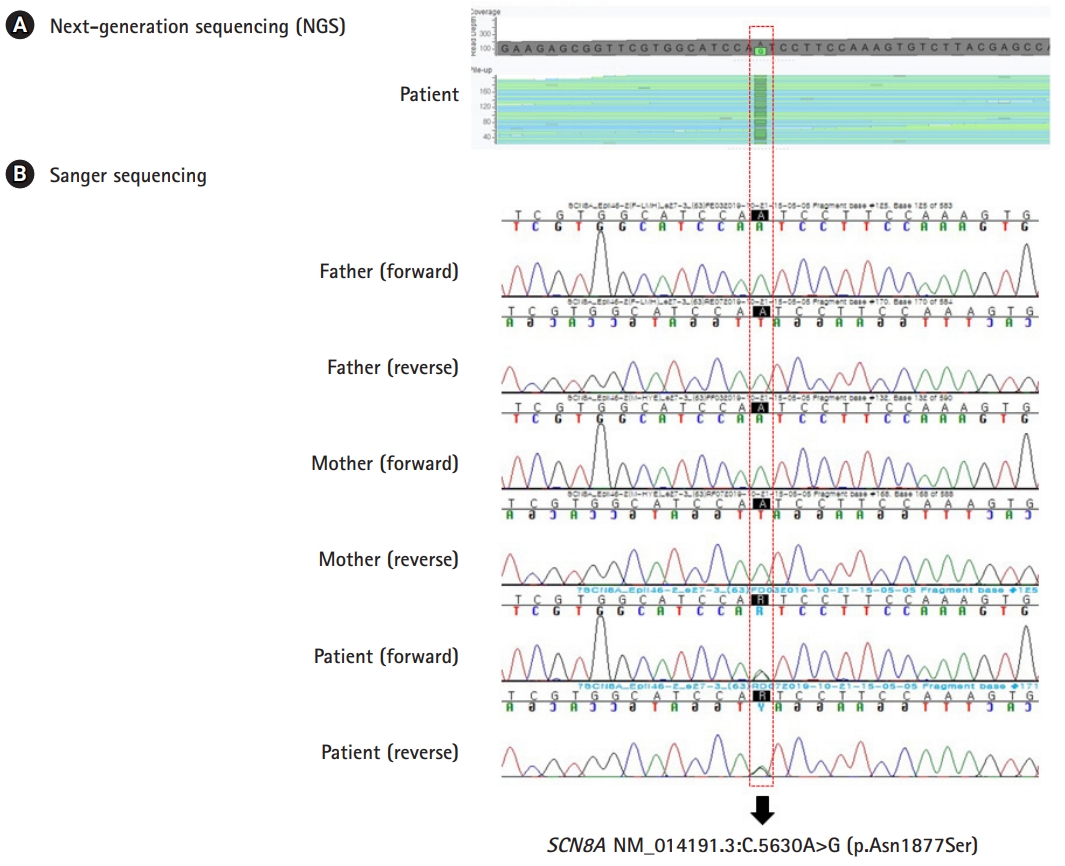
Identification of de novo SCN8A variant. (A) Next-generation sequencing of the patient revealed a heterozygous variant in the SCN8A gene (NM_014191.3:c.5630A>G;p.Asn1877Ser). (B) Sanger sequencing of the patient and both parents confirmed the de novo occurrence of the SCN8A variant.
In general, seizures caused by SCN8A PVs begin in infancy, particularly within 6 months of age. Although focal seizures are the most common, diverse seizure types, including epileptic spasms, atypical absence, myoclonic, and generalized tonic-clonic, can present. Seizures in patients with SCN8A PVs are often controlled effectively by sodium channel blockers, such as phenytoin, carbamazepine, and oxcarbazepine, although other anti-epileptic drugs fail to control seizures [3]. Although infants with SCN8A PVs show normal or mildly delayed development prior to seizure onset, the majority of patients have apparent developmental delay or regression after seizure onset. Motor manifestations, such as hypotonia, dystonia, hyperreflexia, and ataxia can also be presented [3]. Recently, however, Gardella et al. [2] reported that 16 individuals in 3 families with the same missense variant (c.4447G>A, p.Glu1483Lys) in SCN8A gene presented with benign infantile seizures and paroxysmal dyskinesia.
To the best of our knowledge, so far four cases with SCN8A c.5630A>G (p.Asn1877Ser) variant presenting with a mild phenotype, including the present case, were documented (Table 1) [1,4]. However, the same variant presenting with a more severe phenotype of epilepsy and intellectual disability has been also reported [5]. The SCN8A c.5630A>G (p.Asn1877Ser) is located at the proximal 2/3 of the c-terminal domain of Nav1.6 [4]. This is a highly conserved part of the Nav1.6 protein which contains binding sites for interacting proteins. One report speculated that protective genetic variants, or modifier genes which diminish the severity of the SCN8A, could result in a milder phenotype [4].
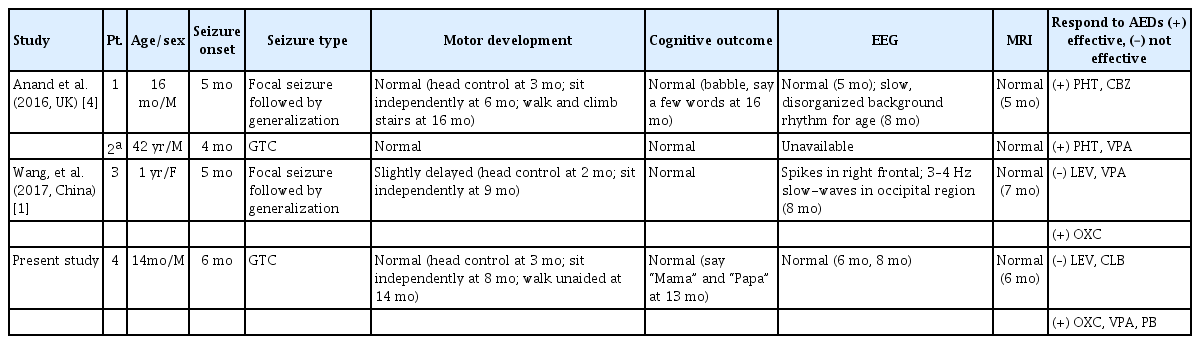
Clinical features in four cases of the SCN8A c.5630A>G (p.Asn1877Ser) variant presenting with a mild phenotype
This case demonstrated that SCN8A c.5630A>G (p.Asn1877Ser) variant can present with relatively mild neurological manifestations. Early genetic testing in patients with refractory seizures with unknown etiology is critical, since identification of epilepsy related to specific gene mutations may help determine the most effective medication, such as sodium channel blockers for SCN8A-related epilepsy, and predict the prognosis.
Notes
No potential conflict of interest relevant to this article was reported.
Author contribution
Conceptualization: KYL. Data curation: EK, CSK, SP, and KYL. Formal analysis: CSK and KYL. Methodology: CSK, SP, and KYL. Visualization: CSK. Writing-original draft: EK and KYL. Writing-review & editing: CSK and KYL.