Ohtahara syndrome is rare and the earliest-appearing age-related epileptic encephalopathy. It is also known as early infantile epileptic encephalopathy with burst-suppression [1]. Seizures onset occurs during the first 3 months of life, often in the form of frequent tonic spasms and partial seizures within the first 2 weeks. We report the first case of Ohtahara syndrome with a novel missense mutation of potassium voltage-gated channel subfamily Q member 2 (KCNQ2) gene (p.Arg207Pro) in Korea that was partially responding to ketogenic diet.
Our patient was born at 38 weeks, 2 days gestation after an uncomplicated pregnancy. Her birth weight was 3.2 kg, and Apgar score was 8/9. She had uncontrollable seizures from the 2nd day after birth. Seizures were tonic in nature involving both the upper limbs and lower limbs, with left deviation of the eyes and cyanosis. Multiple attacks were encountered per day, and each episode took approximately 1 minute. General and neurologic examinations were unremarkable except mild head lagging. Laboratory investigations including electrolyte, congenital infection and test of inborn error of metabolism showed no significant findings. First electroencephalography (EEG), done 6 days after birth, showed hypsarrhythmia and burst-suppression pattern. First magnetic resonance imaging of the brain done on the same day was normal.
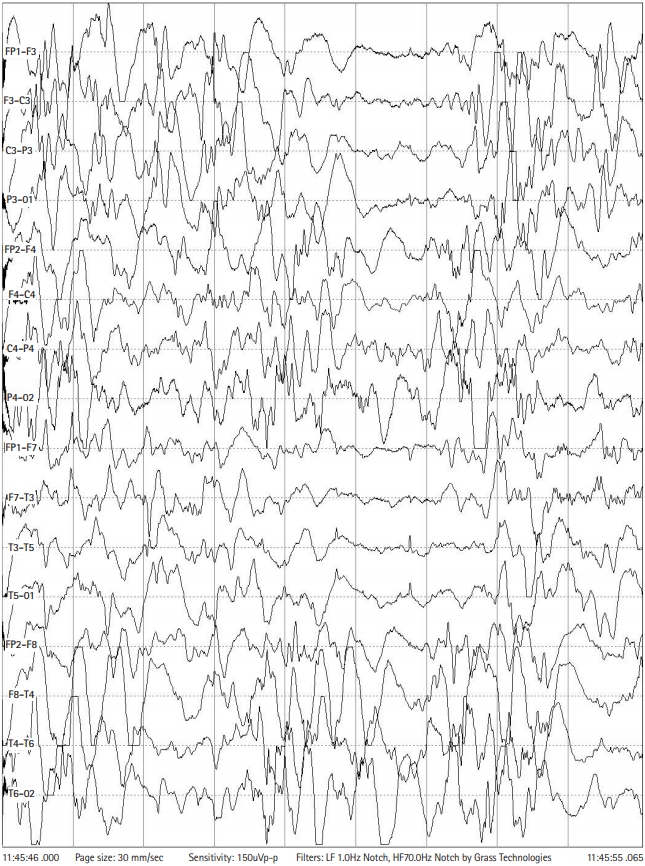
Although multiple antiepileptic drugs were given, brief tonic or myoclonic seizures continued about 10 times a day. Also, her development was constantly delayed. At 6 months of age, her EEG revealed hypsarrhythmia with chaotic background and multifocal sharp waves (Fig. 1). Steroid pulse therapy was not effective. She was therefore put on ketogenic diet at 6 months of age using a ketogenic formula (Ketonia, Namyang Co. Ltd, Seoul, Korea). After a few days of complete ketogenic diet, her seizures were reduced to one or two times a day. Her ketogenic diet continued for 24 months and then gradually stop.
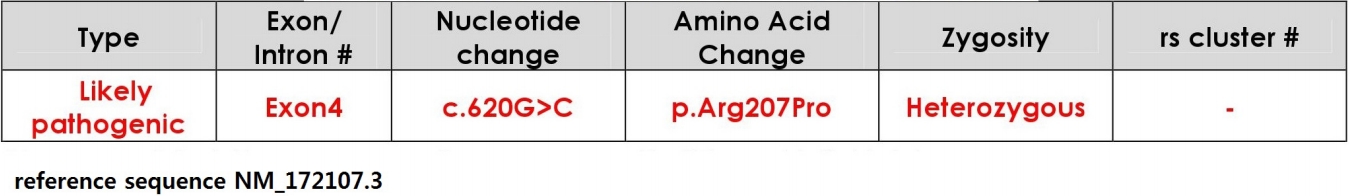
Her genetic test with next generation sequencing revealed a missense mutation in KCNQ2 gene (c.620G>C [p.Arg207Pro]) and were likely pathogenic by American College of Medical Genetics and Genomics guideline 2015, that were not previously reported (Fig. 2). KCNQ2 gene test for her parents showed negative result. Therefore, she was identified as Ohtahara syndrome due to de novo mutation of KCNQ2 gene. Currently, she had brief tonic or myoclonic seizures about less than once a day and bed-ridden state.
Among KCNQ2 related epilepsy, Ohtahara syndrome is the most critical finding for discrimination [2]. Sodium channel blockers like phenytoin and carbamazepine have been reported to be effective in the treatment of KCNQ2-related epilepsy [3]. In this patient, we used phenytoin and topamax but both were not effective at all and seizure was partially controlled by ketogenic diet. Ketogenic diet, mechanistic target of rapamycin inhibitors, is an effective targeted treatment for KCNQ2 encephalopathy, respectively. The patient showed partial response to the ketogenic diet at 6-month-old, but the seizure was not fully controlled and she is currently on a bed-ridden state. Therefore, early and active genetic testing for precision medication is important in patients with suspected Ohtahara syndrome.