Atypical benign partial epilepsy (ABPE) is a complicated form of benign epilepsy with centrotemporal spikes (BECTS) [1]. It is characterized by an earlier age of onset, multiple seizure types including epileptic negative myoclonic, atonic, and atypical absence seizures, as well as focal motor seizures [2]. Electroencephalography (EEG) depicts frequent centrotemporal spikes that are activated during sleep, and similar continuous spike and wave during slow wave sleep (CSWS), but it does not fulfil the standard criteria for CSWS [3]. It has recently been suggested that ABPE is a component of the epilepsy-aphasia spectrum that includes some disorders ranging in severity, where BECTS occurs at the mild end of the spectrum and the severe end includes Landau-Kleffner syndrome and epileptic encephalopathy with CSWS [3]. Patients with ABPE usually exhibit normal neurologic and mental functions through the course, and favorable outcomes [2]. Notably however, some patients reportedly experience cognitive decline, language impairment, and motor skill regression [1,4]. This report describes a boy with ABPE who underwent a very active seizure period accompanied by regression of motor skills, speech, and cognitive function, that was ultimately controlled via ethosuximide and prednisolone.
A 3-year-old boy was referred to our clinic for evaluation of focal motor seizures characterized by jerking of his right arm and leg, facial twitching, and drooling during sleep. He had no remarkable medical history. There was no known family history of epilepsy. His developmental milestones were reportedly normal, which was categorized into peer or high-level group over all areas including gross motor, fine motor, cognition, communication, social interaction, and self-control by the Korean Developmental Screening Test. Physical examination and brain magnetic resonance imaging results were normal. EEG depicted high-voltage and biphasic sharp waves activated by sleep in the left centrotemporal area with normal background (Fig. 1A). The electro-clinical diagnosis was BECTS. Over the following year, while being treated with oxcarbazepine, levetiracetam, and night-time clobazam, facial and lip clonic seizures continued to occur every 1 or 2 weeks. From the age of 4 years he experienced recurrent bilateral tonic-clonic seizures or hemiclonic seizures of the right arm and leg, with Todd paralysis upon waking. Oxcarbazepine, levetiracetam, and clobazam were discontinued because his seizures were controlled after the addition of valproic acid. Interictal EEG depicted typical rolandic spikes. This study was approved by the Institutional Review Board (IRB) of Yonsei University (IRB no., 19-0010). Informed consent was waived due to the retrospective nature of the study.
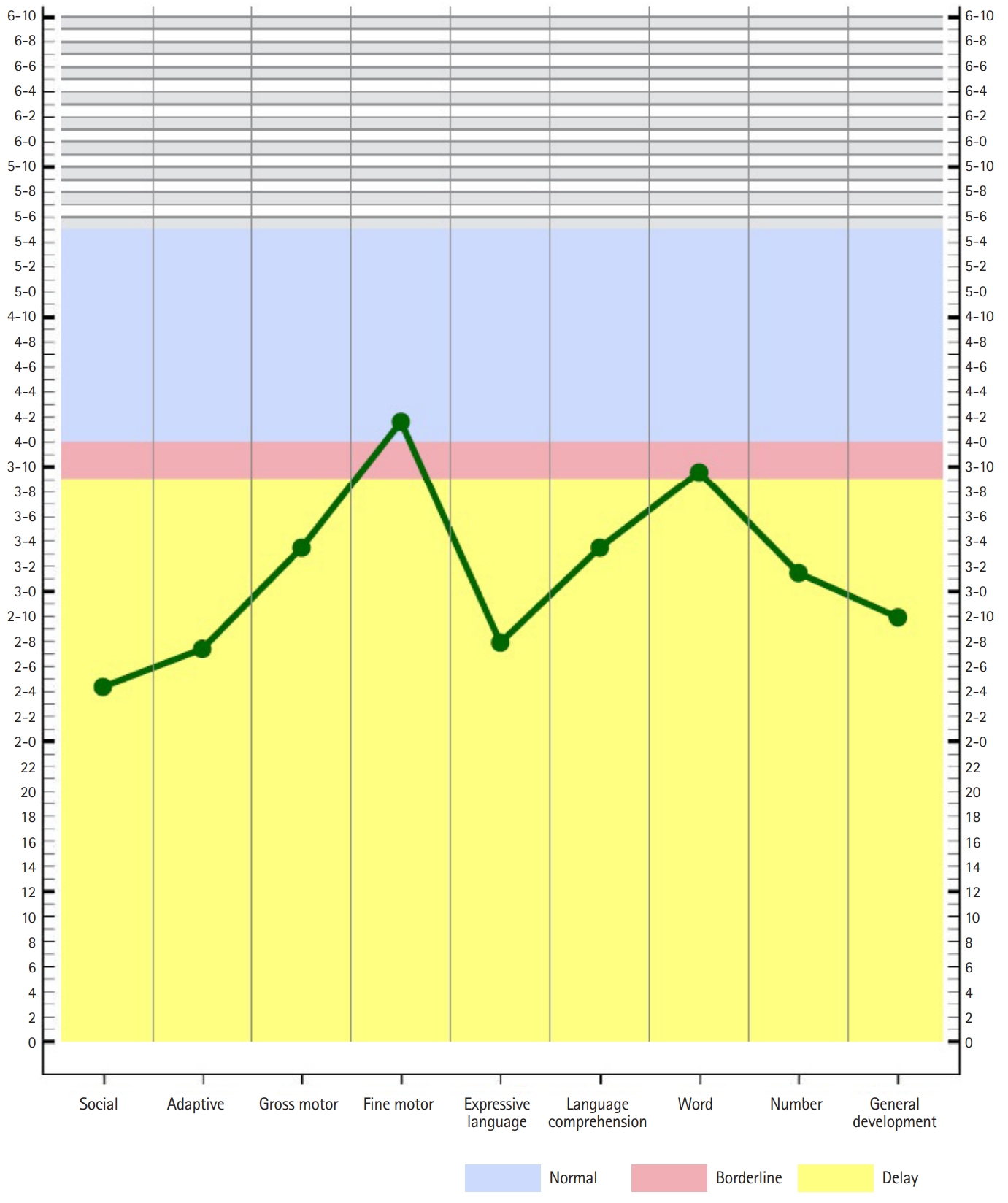
At the age of almost 5.5 years, drop attacks developed with staring, frequent falls, and an unsteady gait. Duration of seizure was short in less than a minute, and there was no postictal drowsiness. He exhibited slurred speech, dysarthria, and dysphagia with drooling. Sleep EEG depicted continuous high amplitude asymmetric spike-and-wave discharges, predominantly in the left centrotemporal area, similar to CSWS (Fig. 1B). Assessment via the Preschool Receptive-Expressive Language Scale and Korean-Child Development Inventory revealed that his development was severely delayed in all domains except for fine motor area (Fig. 2). His equivalent developmental age with regard to receptive language was 43 months, for expressive language it was 40 months, and his integrated developmental age was 2 years and 9 months. Oral prednisolone (2 mg/kg/day) combined with ethosuximide (20 mg/kg/day) was added to valproic acid. His motor skills, language, and seizures started to improve rapidly. Surprisingly, at a follow-up examination 4 weeks later EEG revealed no abnormal epileptic discharges (Fig. 1C). Prednisolone was slowly tapered off, and it was discontinued after 2 months.
At age 8 years, he remains seizure-free with ethosuximide and valproic acid. He continues to yield normal neurologic examination results, and is attending elementary school. EEG depicts focal spikes in the left centrotemporal areas (Fig. 1D). His results in the Korean Wechsler Intelligence Scale for Children (K-WISC-IV) revealed a borderline full-scale intelligence quotient (IQ) of 74 (verbal comprehension 80, perceptual reasoning 91, working memory 70, processing speed 76).
Diagnostic exome sequencing was performed to detect potentially genetic causes of ABPE. The heterozygous missense variant of GABRA1 (NM_000806: c.529G>A) was detected, and was confirmed via direct sequencing. Segregation analysis revealed that the GABRA1 variant was paternally inherited.
In the current patient, clinical seizure history and EEG findings suggest ABPE [3]. Although we could not acquire ictal EEG and electromyography data from the patient, the semiology of seizures such as the sudden drop attacks and frequent staring with no response to stimuli suggest epileptic negative motor and atypical absence seizures, which are primary components of ABPE semiology. EEG depicted sleep activation of focal epileptiform discharges with asymmetric continuous spike-and-wave activity during sleep. The patient exhibited cognitive regression in conjunction with active epilepsy. We did not detect any meaningful genetic variants in the present patient. Recent identification of GRIN2A variants in a small subset of epilepsy-aphasia spectrum patients is notable; however, the associated phenotypic expression is evidently highly variable and may be affected by multiple genes or gene modifiers [1,4].
The current patient’s seizures were resistant to various antiepileptic drugs, but his epilepsy and cognitive function improved after the addition of ethosuximide and prednisolone, which have previously been reported to be effective in ABPE [1,5]. The mechanism by which ethosuximide affects neuronal excitability is considered to involve blockage of T-type calcium channels in thalamic neurons and the corresponding cortex [5]. Therefore, the dramatic effects of ethosuximide may be due to unique antiepileptic effects on epileptic networks in both the thalamus and sensorimotor cortex, but the exact mechanisms remain to be elucidated [5]. In patients who do not respond to antiepileptic drug therapy, corticosteroid administration should be considered in order to prevent language disturbance [1,5]. Steroids and some other immunomodulating therapies are also reportedly effective in ABPE, suggesting that the immune system may be involved in the pathophysiology of ABPE [1,2,5].
In conclusion, ABPE is a distinctive epileptic syndrome characterized by early onset, typical rolandic seizures followed by negative motor seizures, and aggravation to diffuse and continuous epileptic discharges on EEG. Ethosuximide and steroid are effective and safe for the treatment of ABPE, and should be considered relatively early in the course of disease, particularly given the potential for the preservation of intellectual functioning associated with early intervention.